ARTÍCULO ORIGINAL
doi: http://dx.doi.org/10.7705/biomedica.v35i3.2573
1 Grupo de Genética Molecular Humana, Sección de Genética, Departamento de Biología, Universidad del Valle, Cali, Colombia
2 King´s College London, School of Medicine, The James Black Centre, London, England, United Kingdom
Institutions where the research was carried out: This research was carried out at the Laboratorio de Genética
Molecular Humana, Departamento de Biología, Universidad del Valle and the Molecular Hematology Lab, School of Medicine, King´s College London
Author´s contributions: Guillermo Barreto designed and supervised the study. Cristian Fong collected and processed samples, and analyzed and interpreted the data. Stephan Menzel analyzed and interpreted the data. María Alejandra Lizarralde collected and processed samples.
All authors reviewed the final manuscript.
Recibido: 10/10/14; aceptado: 27/04/15
Introduction: Fetal hemoglobin is an important factor in modulating the severity of sickle cell anemia. Its level in peripheral blood underlies strong genetic determination. Associated loci with increased levels of fetal hemoglobin display population-specific allele frequencies.
Objective: We investigated the presence and effect of known common genetic variants promoting fetal hemoglobin persistence (rs11886868, rs9399137, rs4895441, and rs7482144) in 60 Colombian patients with sickle cell anemia.
Materials and methods: Four single nucleotide polymorphisms (SNP) were genotyped by restriction fragment length polymorphisms (RFLP) and the use of the TaqMan procedure. Fetal hemoglobin (HbF) from these patients was quantified using the oxyhemoglobin alkaline denaturation technique. Genotype frequencies were compared with frequencies reported in global reference populations.
Results: We detected genetic variants in the four SNPs, reported to be associated with higher HbF levels for all four SNPs in the Colombian patients. Genetic association between SNPs and HbF levels did not reach statistical significance. The frequency of these variants reflected the specific ethnic make-up of our patient population: A high prevalence of rs7482144-‘A´ reflects the West-African origin of the sickle cell mutation, while high frequencies of rs4895441-‘G´ and rs11886868-‘C´ point to a significant influence of an Amerindian ethnic background in the Colombian sickle cell disease population.
Conclusion: These results showed that in the sickle cell disease population in Colombia there is not a unique genetic background, but two (African and Amerindian). This unique genetic situation will provide opportunities for a further study of these loci, such as fine-mapping and molecular-biological investigation. Colombian patients are expected to yield a distinctive insight into the effect of modifier loci in sickle cell disease.
Key words: Fetal hemoglobin, polymorphism, single nucleotide; anemia, sickle cell; admixture, beta-globin cluster, BCL11A, HBS1L-MYB.
doi: http://dx.doi.org/10.7705/biomedica.v35i3.2573
Las variantes genéticas asociadas con niveles de hemoglobina fetal señalan diversos orígenes étnicos en pacientes colombianos con anemia falciforme.
Introducción. La hemoglobina fetal es un importante factor modulador de la gravedad de la anemia falciforme, cuya expresión está muy condicionada por el factor genético. Los loci asociados con el incremento de la hemoglobina fetal pueden presentar frecuencias alélicas específicas para cada población.
Objetivo. Investigar la presencia y el efecto de las variantes genéticas rs11886868, rs9399137, rs4895441 y rs7482144 asociadas con la persistencia de hemoglobina fetal, en 60 pacientes colombianos con anemia falciforme.
Materiales y métodos. Se hizo la genotipificación de los polimorfismos de nucleótido simple ( Single Nucleotide Polymorphisms, SNP) mediante la técnica de polimorfismos de longitud de fragmentos de restricción ( Restriction Fragment Length Polymorphisms, RFLP) y el procedimiento TaqMan. La hemoglobina fetal (HbF) se cuantificó utilizando la técnica de desnaturalización alcalina de la oxihemoglobina. Las frecuencias genotípicas se compararon con las reportadas en poblaciones de referencia global.
Resultados. Se observaron variantes genéticas ya reportadas para aumento de HbF en los cuatro SNP. La asociación genética entre los SNP y el incremento de la HbF no alcanzó significancia estadística. La frecuencia de estos alelos reflejó la siguiente composición específica en esta muestra de pacientes colombianos: una gran prevalencia de rs7482144-‘A´, lo que indica que el origen de la mutación para la anemia falciforme es África occidental, y una gran frecuencia de rs4895441-‘G´ y rs11886868-‘C´, lo que denota la influencia significativa del origen genético amerindio.
Conclusión. Los resultados evidenciaron que la población con anemia falciforme de Colombia no tiene un único origen genético, sino que existen dos (africano y amerindio). Esta situación genética única ofrece la oportunidad de llevar a cabo un estudio más amplio de estos loci a nivel molecular. Se espera que el estudio de pacientes colombianos permita una visión diferente del efecto de los loci modificadores en esta enfermedad.
Palabras claves: hemoglobina fetal, polimorfismos de nucléotido simple, anemia falciforme, diversidad étnica, globinas beta, BCL11A, HBS1L-MYB.
doi: http://dx.doi.org/10.7705/biomedica.v35i3.2573
Fetal hemoglobin (HbF) ( g 2 a 2 ) is present in human red blood cells from the sixth week of gestation, but is replaced by adult hemoglobin starting from just before birth (1). In some individuals, significant amounts of HbF continue to be produced during adulthood. This is of clinical significance when it happens in patients with beta-thalassemia and sickle cell anemia, resulting in a reduced disease severity and mortality. HbF levels are largely deter-mined by the number of a sub-set of red blood cells termed ‘F cells´ (HbF-containing erythrocytes).
In healthy individuals, F cell levels are largely genetically determined, with a heritability of 89% (2). In beta-globin gene disorders, such as beta- thalassemia and sickle cell disease, F cells and HbF are usually elevated due to stress erythropoiesis, but can be further boosted by common genetic factors. In addition to F-cell levels, factors directly influencing the expression of HBG1 and HBG2 , the genes encoding the specific gamma chain of HbF, contribute to peripheral HbF variability.
Some common genetic variants at quantitative trait loci (QTL) for F cells and HbF have been identified. Sequence variants within the beta-globin gene cluster affect the expression of HBG1 and HBG2 , which encode the gamma chain of fetal hemoglobin, such as rs7482144 (also termed XmnI G g ), a polymorphism within the promoter of HBG2 . Two regions with significant influence on HbF levels have been discovered outside the beta- globin cluster. BCL11A encodes a transcription factor that regulates gamma globin production and SNPs within intronic regulatory sequences that influence HbF levels in healthy individuals and hemoglobinopathy patients. The second region, HBS1L-MYB, contains a series of sequence variants that alter upstream regulative regions for MYB, which encode a transcription factor essential to erythropoiesis. These variants occur in two sub-loci, HMIP-A and HMIP-2B, that map to distinct enhancer elements for MYB (3,4, Menzel, et al. , manuscript in preparation). HbF-increasing alleles at all three regions loci have subsequently been shown to alleviate the severity of sickle cell anemia and beta-thalassemia (5,6). Additional QTLs have been detected on chromosomes 8 and X through linkage analysis but have not been mapped in detail (7). HbF QTLs display population-specific allele frequencies and, hence, their individual importance differs among major population groups and, therefore, in countries.
Sickle cell anemia, a globin disorder mainly of Sub-Saharan African origin, is prevalent in Colombia (8,9), which has a significant African-descendant population component, in addition to European and Amerindian constituents. The significant genetic admixture of Colombian patient groups means that little is known about the presence and frequency of the disease modifier variants at the HbF QTLs.
The present study determined the frequency of variants at the three main known sickle cell disease modifier loci in a sample of patients from Colombia: for rs11886868 at BCL11A gene; for rs9399137 and rs4895441 at HBS1L-MYB; and for rs7482144 at the beta-globin gene cluster. These SNPs have been found to be consistently associated with HbF in diverse populations and groups of patients with sickle cell disease (5,10-13). Our group of 60 patients was not large enough for a formal association analysis, but sufficient to gain insight into the presence of known protective variants as a first step towards a future contribution of genetic research to clinical management and prognosis of patients.
Materials and methods
Sample
Blood samples were collected from 60 individuals (30 female, 30 male) who had been classified as positive for sickle cell hemoglobin (Hb S) (with a metabisulfite test) in Hospital San Jerónimo in Montería (Department of Córdoba), and Hospital Napoleón Franco in Cartagena (Department of Bolívar), both located in the Colombian Atlantic Coast, and Hospital Regional de Buenaventura (Buenaventura) and Hospital Universitario del Valle, HUV (Cali), both in the Colombian Pacific Coast ( Department of Valle del Cauca). Out of those samples, 19 were from the Pacific Coast, and 41 from the Atlantic Coast.
The participant´s age range was from 2 to 45 years. An age cut-off of >2 years was applied so that “adult-type" HbF production would not be confounded with residual “fetal-type" HbF synthesis present during the perinatal period.
Most of the patients were recruited during non-acute visits to the hospitals in the cities where the study took place, and none were in sickle crisis at the time of sampling. The participants had not received blood transfusions during the three months before the samples were taken. The samples were taken after parents or patients gave written informed consent. This research was approved by the ethic board of the Universidad del Valle (Approval act 09-09).
Quantification of HbF in whole blood
HbF was quantified using the oxyhemoglobin alkaline denaturation technique (14, 15). For this, all hemoglobin, except HbF, present in the hemolysate underwent denaturation in an alkaline medium. After the precipitation and filtration of this hemolysate, the HbF recovered was quantified by spectrophotometry at 450nm.
Molecular diagnosis of HbS
DNA was extracted using the salting-out technique (16). The genotype at the HbS locus was determined using a PCR-RFLP assay, as described in one of our previous studies (17). This method of diagnosis for sickle cell anemia made possible to exclude patients with heterozygous genotype such as HbSC or HbS b thal + ; however, patients with HbS/ b thal 0 genotype due to beta gene deletion could not be excluded.
SNP genotyping
For SNP genotyping of rs9399137, rs4895441 and rs11886868, we used TaqMan assays (18) (Applied Biosystem, Foster City, California) run on a TaqMan 7900 real-time PCR instrument (Applied Biosystems) with supplier-recommended procedures. For SNP rs7482144, a restriction digestion with Xmn I was carried out after PCR. The primers used for this region were Forward 5´CAGGCCTCACTGGAGCTACAGAC and Reverse 5´ACCTCAGACGTTCCAGAAGCGAGT, which had been designed using the PRIMER3 software (19). The PCR mix contained 50 ng of genomic DNA in a 25 l volume with 0,12 uM each primer, 0.01 mM total of dNTP´s 1X TRIS-HCl, pH 8.4, 50 mM KCl, 1 mM MgCl2 and 1unit of Taq polymerase (Bioline, London, United Kingdom). The PCR conditions were as follows: Initial denaturation at 92° for 5 min, 30 cycles with denaturation at 93° for 1 min, annealing at 68° for 1 min and extension at 72 for 1.5 min and a final cycle extension was prolonged to 3 min.
Data analysis
Observed frequencies were compared with those reported by HapMap for reference populations and The 1000 Genomes Project (20,21). The association of SNP alleles with HbF levels was evaluated by linear regression, with age and sex as covariates. Association was also tested with a joint ‘SNP score´ (22). Allele frequencies between populations were compared using chi-square test. All analysis were performed using Statistica 8.0 (23) and Arlequin software (24).
Results
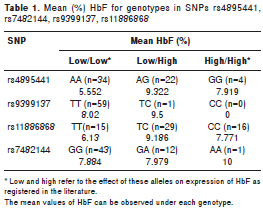
We genotyped representative SNPs for four fetal-hemoglobin QTLs (BCL11A, HMIP-2A, HMIP-2B, the beta-globin gene cluster) in 60 Colombian patients with sickle cell anemia. Alleles known to increase HbF levels in European and West African populations were detected at each locus. Patients carrying such alleles (heterozygotes ) had, on average, higher HbF levels than patients without these alleles, for all four SNPs investigated (table 1), however, a formal test for genetic association did not reach statistical significance (in order to get it, we would probably have to increase the number of patients studied). Age was the only variable significantly associated with HbF (p=0.014) (result not shown).
The frequency of HbF-promoting alleles across the four loci is strikingly different (table 2) from that reported for West African, European and East Asian populations: The first QTL, the beta-globin gene cluster, is represented by rs7482144 (also called ‘Xmn I G g site´). This SNP is linked to the sickle mutation, and allele ‘A´ largely indicates the presence of the ‘Senegal´ sickle haplotype, known to be associated with higher HbF levels and milder disease. ‘A´ is somewhat more frequent in our patients than in the West African reference population YRI (0.123 vs. 0.074, not significant). This allele did not show differences with HCB population either (0.123 vs 0.113).
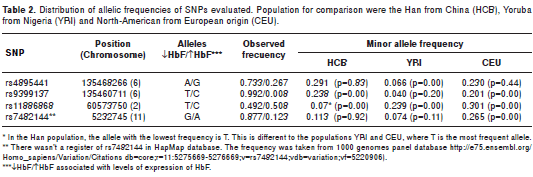
The BCL11A locus is represented by SNP rs11886868, where the high-HbF (and protective) allele is ‘C´ in all populations studied so far (22,25-27). In the Colombian samples, the two alleles have a nearly equal frequency (T=0.492 and C=0.508) (table 2). This is fundamentally different to what has been reported for West Africans or Europeans, where ‘C´ is the minor allele (with frequencies of 0.239 and 0.301, respectively), but also for East Asians (Han Chinese, HCB), where ‘C´ is predominant (at 0.930). Thus, Colombian patients seem to represent a situation at this locus where an ‘East-Asian like´ component is overlaid over West-African/European allele frequencies.
The HBS1L-MYB region contains two HbF QTLs, HMIP-2A and HMIP-2B (HBS1L-MYB intergenic polymorphisms A and B) (Menzel, et al. , manuscript in preparation). HMIP-A is tagged by the SNP rs9399137, with the ‘C" allele promoting HbF. This allele is very rare in the Colombian patients, carried only by a single individual; rs9399137-‘C´ is also rare in the West-African YRI population sample (0.04), but significantly more common (p<0.01) in Chinese and Europeans. HMIP-B was tagged in our patients by rs4895441, where the HbF-promoting ‘G´ allele was significantly more common (0.267 vs. 0.066, p<0.05) than in the West African (YRI) population, but similar in frequency to the Chinese (HCB, 0.29) or European (CEU, 0.23) populations. The presence of rs4895441 -‘G ´ in individuals that lack rs9399137-‘C´ (a common situation in Colombian patients) is untypical for Europeans though, but characteristic in some East-Asian and Amerindian populations (Menzel, et al ., manuscript in preparation).
Discussion
We investigated four major QTLs affecting HbF levels (the beta-globin gene cluster, BCL11A, HMIP-2A, and HMIP- 2B) in 60 Colombian patients with sickle cell disease. While our patient group was too small to formally show association with representative markers for these loci, we evaluated the frequency of HbF-promoting alleles to obtain a first overview of the genetic situation of patients with sickle cell disease in Colombia.
The SNPs evaluated in this paper showed a strong association with the HbF levels in a wide spectrum of populations, including those from Europe, North America and Brazil where three joint QTLs (rs9399137, 4895441 y rs11886868) explain more than 20% of HbF variability (5,18,22 ). Therefore, they are suited to estimate the prevalence of HbF-promoting alleles at these loci.
Genotypes for rs7482144 represent West-African segments of chromosome 11 containing the beta-globin locus and sickle mutation. The prevalence of allele ‘A´, tagging mostly the ‘Senegal´ sickle haplotype and a few atypical haplotypes (28), is due to the specific origin of the African ancestors of our patients, which might have included individuals from Senegal or Sierra Leone, where this allele is more frequent (29).
Alleles at the two regions unlinked to the sickle mutation, BCL11A and HBS1L-MYB (HMIP-A and HMIP-2B), showed allele frequencies that are distinctly non-African, suggesting a substantial influence of admixture in the genetic situation of sickle patients in Colombia.
At BCL11A, the high frequency of the rs11886868-‘C´ high-HbF allele suggests the presence of a large East-Asian-like population component. This component is most likely of Amerindian nature since substantial similarities exist between some East Asian and Amerindian population groups (30).
At HMIP-2A, the high-HbF rs9399137-‘C´ allele was present in a single patient. This patient also carried rs4895441-‘G´ at HMIP-2B, indicating the presence of a ‘Eurasian haplotype´ (Menzel, et al ., manuscript in preparation), which is not present in West Africa, but is likely to originate from Asian , European or Amerindian chromosomes; rs4895441-‘G´, on its own, was relatively frequent in our patients. This situation is characteristic for some East Asian, such as Japanese and Yakut, and Amerindian populations (Human Genome Diversity Panel (http://www.hagsc.org/hgdp/files.html).
These overall results are probably an indication of the genetic diversity shown by the Colombian population. The Colombian Amerindian population component is genetically complex, with large differences in allelic frequency among sub-populations, possibly due to a strong genetic drift and small effective population sizes (31). In the pre-Columbian period, there were several Amerindian populations that maintained isolation from each other inhabiting the territory of today´s Colombia (32). In the present population from the Center and the South-West of Colombia, the presence of admixture and gene flow between different ethnical groups is well documented. Using markers for mtDNA, Rondón, et al ., (33) showed that in individuals with African heritage, there were 15% typical Amerindian haplotypes. In addition, complex admixture patterns have been observed in other population groups, such as indigenous and “mestizo" (i. e., of mixed descent) groups in the South-West of Colombia, but also in the European/Amerindian admixed population of the Department of Antioquia (North-West of Colombia). This popu lation showed a high frequency of Amerindian haplotypes in mtDNA (77-100%). Regarding the Y chromosome, 94% was European, 5% African and 1% Amerindian. These data suggest a model of admixture for this part of Colombia: Predominantly male European founders from Southern Spain and a female predominantly Amerindian contribution followed by isolation (34,35). Thus, the admixture between European, African, and Amerindian populations has a different extent in each region of Colombia. In the center and East of Colombia, European and Amerindian ancestry predominate; in coastal regions, European, Amerindian, and African ancestry all significantly contribute to the population (36). Specifically, our data indicate the presence of a large Amerindian admixture component in our patients, resulting in a comparatively high prevalence of HbF-promoting and disease-alleviating QTL alleles.
The results of this study show that sickle cell anemia patients have an appreciable admixture between people from African and Amerindian origins. This phenomenon is typical in Colombian population, and is the product of historical events that gave rise to it (37). When the Spaniards came to the actual Colombian territory, the Indian groups at both coasts were Arawaks and Caribes on the Caribbean Coast; and Embera, Weunana, and Cuna on the Pacific Coast (38). The first Africans arrived in America in 1525. This first group of slaves came from Senegal and Sierra Leone (Yolofo, Serere group) (39,40,41). The origin of these first slaves could explain the prevalent frequency of Senegal allele at rs7482144. Subsequent organization of Afro Colombian communities in Palenques (isolated settlements of free people) allowed the breeding with Amerindian people present on coastal regions of Colombia. This admixture accounts for the observed allele frequency of SNPs assessed in this study.
An extension of studies reported here will require the enrollment of additional patients, enabling a detailed investigation of the relationship between QTL polymorphisms and HbF. Amerindian genes have not been studied extensively in connection with this trait, and different SNPs might be best suitable to tag HbF-promoting alleles with this specific ancestry. With their unique genetic back-ground, Colombian patients are expected to yield a distinctive insight into the effect of modifier loci in sickle cell disease.
We wish to thank Professor Swee Lay Thein, head of Molecular Hematology at King´s College, London, for hosting Cristian Fong in her lab, where he conducted part of the genetic experiments of this study. To all staff of Hospital Regional de Buenaventura, Hospital Universitario del Valle in Cali, Hospital San Jerónimo in Montería, Hospital Infantil Napoleón Franco in Cartagena, and to all participants.
The authors declare that they have no conflicts of interest in the research.
Contract grant sponsor: Colciencias. Contract grant number: RC-511-2009. Code: 1106-493-26235.
Corresponding author: Guillermo Barreto, Departamento de Biología, Universidad del Valle, Calle 13 N° 100-00, edificio 320, oficina 4040, Cali, Colombia Telephone: (572) 321 2152; Fax: (572) 339 3243guillermo.barreto@correounivalle.edu.co
1. Bank A . Regulation of human fetal hemoglobin: New players, new complexities. Blood. 2006;107:435-43.
2. Garner C, Tatu T, Reittie JE, Littlewood T, Darley J, Cervino S, et al . Genetic influences on F cells and other hematologic variables: A twin heritability study. Blood. 2000;95:342-6.
3. Stadhouders R, Thongjuea S, Andrieu-Soler C, Palstra R-J, Bryne JC, van den Heuvel A, et al . Dynamic long-range chromatin interactions control Myb proto-oncogene transcription during erythroid development. The EMBO Journal. 2011;31:986-99.
4. Menzel S, Jiang J, Silver N, Gallagher J, Cunningham J, Surdulescu G, et al . The HBS1L-MYB intergenic region on chromosome 6q23.3 influences erythrocyte, platelet, and monocyte counts in humans. Blood. 2007;110:3624-6.
5. Lettre G, Sankaran VG, Bezerra MAC, Araújo AS, Uda M, Sanna S, et al . DNA polymorphisms at the BCL11A, HBS1L-MYB, and beta-globin loci associate with fetal hemoglobin levels and pain crises in sickle cell disease. Proc Nat Acad Sci USA. 2008;105:11869-74.
6. Galanello R, Sanna S, Perseu L, Sollaino MC, Satta S, Lai ME, et al . Amelioration of Sardinian 0 thalassemia by genetic modifiers. Blood. 2009;114:3935-7.
7. Fertrin KY, Costa FF. Genomic polymorphisms in sickle cell disease: Implications for clinical diversity and treatment. Expert Rev Hematol. 2010;3:443-58.
8. Silva R, Malambo D, Silva D, Fals E, Fals O, Rey J. Tamizaje de hemoglobinopatías en muestra de la población infantil de Cartagena. Pediatría. 1998;33:86-9. Entry date: August 20, 2013. Available from: http://www.encolombia.com/medicina/pediatria/33-2_pediatria_tamizaje.
9. De Bernal M, Collazos A, Bonilla RD, Tascón EP . Determination of the prevalence of hemoglobin S, C, D, and G in neonates from Buenaventura, Colombia. Col Med. 2010;41:141-7.
10. Danjou F, Anni F, Perseu L, Satta S, Dessi C, Lai ME, et al . Genetic modifiers of thalassemia and clinical severity as assessed by age at first transfusion. Haematologica. 2012;97:989-93.
11. Alsultan A, Solovieff N, Aleem A, AlGahtani FH, Al- Shehri A, Osman ME, et al . Fetal hemoglobin in sickle cell anemia: Saudi patients from the Southwestern province have similar HBB haplotypes but higher HbF levels than African Americans. Am J Hematol. 2011;86:612-4.
12. Galarneau G, Palmer CD, Sankaran VG, Orkin SH, Hirschhorn JN, Lettre G. Fine-mapping at three loci known to affect fetal hemoglobin levels explains additional genetic variation. Nat Genet. 2010;42:1049-51.
13. Thein SL, Menzel S, Peng X, Best S, Jiang J, Close J, et al . Intergenic variants of HBS1L-MYB are responsible for a major quantitative trait locus on chromosome 6q23 influencing fetal hemoglobin levels in adults. Proc Nat Acad Sci USA. 2007;104:11346-51.
14. Sáenz G, Moreira J. Laboratorio hemoglobinopatías. Manual latinoamericano. San José de Costa Rica. 1980. p. 102-104.
15. Sáenz G. Hematología teórico-práctica. Morfología y bio-química hematológica. 7ª edición. 1981. p. 441-44.
16. Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215.
17. Ortega DC, Fong C, Cárdenas H, Barreto G. Evidence of over dominance for sickle cell trait in a population simple from Buenaventura, Colombia. Int J Gent Mol Biol. 2015; 7:1-7.
18. Creary LE, Ulug P, Menzel S, McKenzie CA, Hanchard NA, Taylor V, et al. Genetic variation on chromosome 6 influences F cell levels in healthy individuals of African descent and HbF levels in sickle cell patients. PLoS ONE. 2009;4:e4218.
19. Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, et al. Primer3 - new capabilities and interfaces. Nucleic Acids Res. 2012;40:e115-5.
20. Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, Gibbs RA, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851-61.
21. McVean GA, Altshuler DM, Durbin RM, Abecasis GR, Bentley DR, Chakravarti A, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;490:56-65.
22. Uda M, Galanello R, Sanna S, Lettre G, Sankaran VG, Chen W, et al. Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of beta-thalassemia. Proc Nat Acad Sci USA. 2008;105:1620-5.
23. StataCorp. Stata Statistical Software: Release 11. College Station, TX: StataCorp LP; 2009.
24. Excoffier L, Laval G, Schneider S. Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform Online. 2005;1:47-50.
25. Makani J, Menzel S, Nkya S, Cox SE, Drasar E, Soka D, et al. Genetics of fetal hemoglobin in Tanzanian and British patients with sickle cell anemia. Blood. 2011;117:1390-92.
26. Borg J, Papadopoulos P, Georgitsi M, Gutiérrez L, Grech G, Fanis P, et al. Haploinsufficiency for the erythroid transcription factor KLF1 causes hereditary persistence of fetal hemoglobin. Nat Genet. 2010;42:801-5.
27. Sedgewick AE, Timofeev N, Sebastiani P, So JCC, Ma ESK, Chan LC, et al. BCL11A is a major HbF quantitative trait locus in three different populations with b -hemoglobinopathies. Blood Cells Mol Dis. 2008;41: 255-8.
28. Fong C, Lizarralde-Iragorri MA, Rojas-Gallardo D, Barreto G. Frequency and origin of haplotypes associated with the beta-globin gene cluster in individuals with trait and sickle cell anemia in the Atlantic and Pacific coastal regions of Colombia. Genet Mol Biol. 2013;36:494-7.
29. Pagnier J, Mears JG, Dunda-Belkhodja O, Schaefer- Rego KE, Beldjord C, Nagel RL, et al. Evidence for the multicentric origin of the sickle cell hemoglobin gene in Africa. Proc Natl Acad Sci USA. 1984;81:1771-3.
30. Li JZ, Absher DM, Tang H, Southwick AM, Casto AM, Ramachandran S, et al. Worldwide human relationships inferred from genome-wide patterns of variation. Science. 2008; 319:1100-4.
31. Mesa NR, Mondragón MC, Soto ID, Parra MV, Duque C, Ortiz-Barrientos D, et al. Autosomal, mtDNA, and Y-chromosome diversity in Amerinds: Pre-and post- Columbian patterns of gene flow in South America. Am J Hum Genet. 2000;67:1277-86.
32. Wang S, Ray N, Rojas W, Parra MV, Bedoya G, Gallo C. Geographic patterns of genome admixture in Latin American Mestizos. PLoS Genet. 2008;4:e1000037.
33. Rondón F, Orobio RF, Braga YA, Cárdenas H, Barreto G. Estudio de diversidad genética de cuatro poblaciones aisladas del centro y suroccidente colombiano. Salud UIS 2006;38:12-20.
34. Bedoya G, Montoya P, García J, Soto I, Bourgeois S, Carvajal L, et al. Admixture dynamics in Hispanics: A shift in the nuclear genetic ancestry of a South American popu- lation isolate. Proc Nat Acad Sci USA. 2006;103:7234-9.
35. Carvajal-Carmona LG, Soto ID, Pineda N, Ortiz-Barrientos D, Duque C, Ospina-Duque J, et al. Strong Amerind/ white sex bias and a possible Sephardic contribution among the founders of a population in northwest Colombia. Am J Hum Genet. 2000;67:1287-95.
36. Rojas W, Parra MV, Campo O, Caro MA, Lopera JG, Arias W, et al. Genetic make-up and structure of Colombian populations by means of uniparental and biparental DNA markers. Am J Phys Anthropol. 2010;143:13-20.
37. Rodas C, Gelvez N, Keyeux G. Mitochondrial DNA studies show asymmetrical Amerindian admixture in Afro-Colombian and Mestizo populations. Hum Biol. 2003;75:13-30.
38. Centro de Investigación y Educación Popular. Colombia país de regiones. Santafé Bogotá: Santafe de Bogotá: Cinep: Colciencias; 1998. p. 312.
39. Sánchez-Mejia H. Esclavitud, zambaje, “rochelas" y otros excesos en la población libre de las gobernaciones de Santa Marta y de Cartagena 1600-1800. En: Chicangana-Bayona YA, editor. Historia, cultura y sociedad colonial siglos XVI- XVII. Formas, problemas y perspectivas en Colombia. Colombia: La Carreta editores E.U. Universidad Nacional sede Medellín; 2008. p. 127-57.
40. Maya-Restrepo LA. Demografía histórica de la trata por Cartagena 1533-1810. En: Maya A, coordinador. Geografía humana de Colombia. Los afrocolombianos. Santa Fe de Bogotá: Instituto Colombiano de Cultura Hispánica; 1998. p. 11-52.
41. Navarrete M. Cimarrones y palenques en las provincias al norte del Nuevo Reino de Granada S. XVII. Fronteras de la Historia. 2001; 6:97-122.