Biomédica 2017;37:250-9
doi: http://dx.doi.org/10.7705/biomedica.v37i3.2980
ARTÍCULO ORIGINAL
1 Instituto de Ortopedia Infantil Roosevelt, Bogotá, D.C., Colombia
1 Facultad de Medicina, Departamento de Morfología, Maestría de Genética Humana, Universidad Nacional de Colombia, Bogotá, D.C., Colombia
Contribución de los autores:
Ambos autores contribuyeron a la recopilación, el análisis y la interpretación de los datos, y en la escritura del manuscrito.
Recibido: 16/07/15; aceptado: 07/09/16
Introducción. La prevalencia de talla baja en Colombia es de 10 %, aproximadamente. En el 2009, la International Skeletal Dysplasia Society incluyó 456 condiciones clínicas en su clasificación, con base en criterios bioquímicos, radiológicos y moleculares para su diagnóstico.
Objetivo. Analizar las variables demográficas, epidemiológicas y clínicas en un grupo de pacientes con enfermedades genéticas del esqueleto, remitidos al Instituto de Ortopedia Infantil Roosevelt. Materiales y métodos. Se analizaron pacientes remitidos entre el 2008 y el 2014, con 167 diagnósticos relacionados con enfermedades genéticas del esqueleto según la Clasificación Internacional de Enfermedades, versión 2010 (CIE-10). Se exploraron las variables demográficas, epidemiológicas y clínicas empleando estadística descriptiva. Se generó una puntuación para las intervenciones que contempló las combinaciones de tratamientos, y se analizaron las variables mediante la prueba t de Student.
Resultados. El motivo de consulta más frecuente fue por sospecha de enfermedad genética del esqueleto. Entre los tipos de tratamiento, se consideraron los de soporte, los quirúrgicos, el farmacológico y la ‘ortesis’, y se pudo establecer que los pacientes con enfermedades genéticas del esqueleto obtenían puntajes mayores en la variable de intervención y menores en las de talla alta y baja.
Conclusiones. El diagnóstico de la mayoría de los pacientes remitidos respondía a enfermedades genéticas del esqueleto, talla baja y otras enfermedades genéticas monogénicas. Se encontraron diferencias significativas entre la edad de inicio de los síntomas y la de diagnóstico, así como diversos enfoques terapéuticos. Hubo menos intervenciones en los pacientes con talla alta y baja, lo cual podría alertar sobre la necesidad de reevaluar las necesidades terapéuticas de este grupo.
Palabras clave: enfermedades del desarrollo óseo; esqueleto; genética; estatura; Colombia.
doi: http://dx.doi.org/10.7705/biomedica.v37i3.2980
Introduction: Short height in Colombia has an estimated prevalence of 10%. The 2009 Nosology and Classification of Skeletal Genetic Diseases described 456 clinical conditions using biochemical, molecular and radiological criteria for diagnosis.
Objective: To analyze demographic, epidemiological and clinical variables in a group of patients with skeletal genetic diseases referred to the Instituto de Ortopedia Infantil Roosevelt.
Materials and methods: Patients referred between 2008 and 2014 were analyzed filtering 167 diagnoses of the International Classification of Diseases, 10th revision (ICD 10), related to skeletal genetic diseases. Demographic, epidemiological and clinical variables were explored using descriptive statistics. An intervention score was generated contemplating different combinations of treatments. An inferential statistical analysis using Student’s t test was performed on such variables.
Results: The most frequent reason for consultation was suspicion of a genetic skeletal disorder. The types of treatments considered included support, surgical, pharmacological and orthotics, and it was established that genetic skeletal disorders were associated with higher intervention scores while tall and short height showed a lower score.
Conclusions: Most referred patients were classified with genetic bone diseases, short stature and other monogenic genetic diseases. Significant differences were found between the age at symptoms onset and the age of diagnosis. Diversity was found in the therapeutic approach among different groups of pathologies. Patients with tall and short height showed lower intervention scores, which may warn on the need to reassess the therapeutic requirements of these groups.
Key words: Bone diseases, developmental; skeleton; genetics; body height; Colombia.
http://dx.doi.org/10.7705/biomedica.v37i3.2980
La talla baja tiene una prevalencia en la población general de 2 a 5 % (1,2). Según la Organización Mundial de la Salud (OMS), la prevalencia de niños con déficit de altura ha venido reduciéndose. En Suramérica, esta tendencia también se ha reflejado en la reducción de su frecuencia de 25,1 a 9,3 %, entre 1980 y 2000 (3). En el 2005, la prevalencia de talla baja reportada en Colombia fue de 13,9 %, aproximadamente, sin discriminar según su etiología (4), en tanto que se redujo a 10 % en el 2010 (5).
Esta condición es una de las preocupaciones más frecuentes en los servicios de salud pediátricos de endocrinología, gastroenterología y ortopedia, entre otras especialidades (6,7), y es la segunda causa de consulta en genética en Colombia (8 % de los pacientes atendidos) (8). La talla baja se define clínicamente como una estatura por debajo de -2 desviaciones estándar, o menor al percentil 3, para la edad y el sexo, con respecto a la población general (1,9).
Existen múltiples clasificaciones de la talla baja, por ejemplo, la división entre no patológica (talla baja familiar y constitucional) y la patológica (10). La European Society for Paediatric Endocrinology (ESPE) subdivide esta última en tres categorías: falla primaria del crecimiento, falla secundaria del crecimiento y talla baja idiopática (11). En la primera categoría, se describen los síndromes clínicamente definidos, el grupo de pequeños para la edad de gestación con fallas en el patrón de recuperación del crecimiento y las displasias esqueléticas.
A partir de la creación de la International Skeletal Dysplasia Society (ISDS) en 1999, se constituyó un comité ad hoc encargado de la estandarización clínica, radiológica y molecular de estas displasias esqueléticas (12,13). En la reunión de expertos de agosto de 2009 se elaboró la nosología y la clasificación de las enfermedades genéticas del esqueleto, describiendo 456 condiciones clínicas organizadas en 40 grupos, con base en criterios bioquímicos, radiológicos y moleculares, con el fin de establecer una lista de referencia que sirviera de respaldo en el proceso de diagnóstico (14).
Dadas las limitaciones de información regional y local sobre algunas variables clínicas de los pacientes con talla baja y enfermedades genéticas del esqueleto, el objetivo de este estudio fue analizar dichos elementos en un grupo poblacional, mayoritariamente de niños, remitidos a un centro especializado en enfermedades osteomusculares, y compararlos con algunos de los reportados en la literatura científica mundial.
Materiales y métodos
Se hizo un estudio retrospectivo en el cual se analizaron los pacientes remitidos entre el 2008 y el 2014 a consulta externa y hospitalización en esta institución. Como filtro inicial, se tomaron 167 diagnósticos de la CIE-10 relacionados con enfermedades genéticas del esqueleto, con lo cual se seleccionaron 3.204 historias clínicas; de estas, se escogieron las de aquellos pacientes que acudieron a valoración genética, lo cual resultó en un número de 477 pacientes, cuyos registros se revisaron y se encontraron 35 con datos incompletos, por lo cual no se consideraron para el estudio. Por consiguiente, en total, se incluyeron 442 pacientes.
Se encontraron 289 motivos diferentes de consulta, difíciles de enmarcar en alguna de las clasificaciones clínicas mencionadas y, por ello, se establecieron siete categorías discrecionales para agruparlos y así facilitar el análisis:
1) hallazgos clínicos no relacionados con la talla,
2) otros problemas esqueléticos,
3) sospecha de cromosomopatía,
4) sospecha de enfermedad genética del esqueleto,
5) sospecha de otras enfermedades genéticas monogénicas,
6) talla alta, y
7) talla baja.
Además, se estableció una puntuación para los tipos de intervención, que contempló las combinaciones de tratamientos en una ponderación discrecional (ninguno=0; soporte=1; ‘ortesis’=1; farmacológico=2; quirúrgico=5). También, se valoró el uso de estudios diagnósticos específicos para la enfermedad sospechada, su posible mecanismo de herencia y la presencia de antecedentes familiares asociados.
Posteriormente, para una mejor comprensión de sus condiciones y para establecer relaciones entre las variables analizadas, se caracterizó a la población mediante las siguientes técnicas de análisis exploratorio de datos:
a) análisis estadístico unidimensional con cuadros de salida para las variables de sexo, edad de inicio de síntomas y de diagnóstico, motivo de consulta, diagnóstico y tratamiento;
b) análisis estadístico bidimensional, con el cual se relacionaron las diferentes variables del estudio (sexo y edad, edad y motivos de consulta, diagnóstico y cirugías); además, se hicieron pruebas de diferencia de medias entre la agrupación de los pacientes por motivo de consulta y por diagnóstico, y
c) análisis de correspondencias simples para determinar la homogeneidad entre las variables y las agrupaciones de los pacientes según los diagnósticos y los tratamientos (motivo de consulta y diagnóstico final, diagnóstico final y la puntuación de la intervención), mediante la prueba de ji al cuadrado de Pearson, con el fin de analizar la correlación entre estas variables. Los análisis estadísticos se hicieron con el programa SPSS®, versión 22.
Consideraciones éticas
El manejo de la información obtenida de la base de datos del Instituto de Ortopedia Infantil Roosevelt obedeció a estándares de confidencialidad y privacidad. Este trabajo fue debidamente aprobado por el Comité de ética e Investigación de esta institución.
Resultados
Se analizaron 442 individuos, 198 mujeres (44,7 %) y 244 hombres (55,2 %), provenientes en su gran mayoría de Cundinamarca (80,5 %) y, específicamente, de Bogotá (74,4 %); otros sitios de remisión fueron los departamentos de Tolima (5,2 %) y Boyacá (2,7 %).
La condición mayoritariamente diagnosticada en esta población por los médicos consultados fue el enanismo (16,9 %) (código E34.3 de la CIE- 10: “no clasificado en otra parte”), seguido por síndromes de malformaciones congénitas asociadas principalmente con estatura baja (16,4 %) (Q871 de la CIE-10), otros trastornos del desarrollo y neurofibromatosis (9,93 %) (Q850 en la CIE-10).
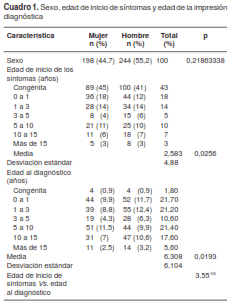
La media de la edad de inicio de los síntomas fue de 2,58 años (intervalo de confianza, IC: ± 4,88). La mayoría de los pacientes presentó los síntomas desde el momento del nacimiento (42,7 %), grupo seguido por la aparición de síntomas entre 0 y 1 año de edad (18 %), y entre 1 y 3 años (14 %) (cuadro 1). En la historia clínica de 15,3 % de los pacientes, se reportaba la aparición de síntomas prenatales y no se encontraron diferencias entre los sexos con relación a este ítem (no se presentan los datos).
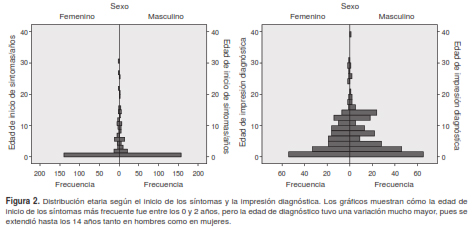
La media de edad de la impresión diagnóstica fue de 6,3 años (IC: ± 6,1). En el 7 % de los pacientes el diagnóstico se produjo en los 12 primeros meses de vida, en el 32 %, entre el primero y el quinto año de vida, y en el 44,6 %, a partir de los 5 años (cuadro 1). En la figura 1 se presenta el amplio espectro de frecuencia de los síntomas e impresiones diagnósticas en todas las franjas etarias analizadas. Al comparar la edad de inicio de los síntomas con la de la impresión diagnóstica, se observó una gran variabilidad entre ellas (figura 2), con diferencias significativas entre las edades promedio de una y otra (cuadro 1).
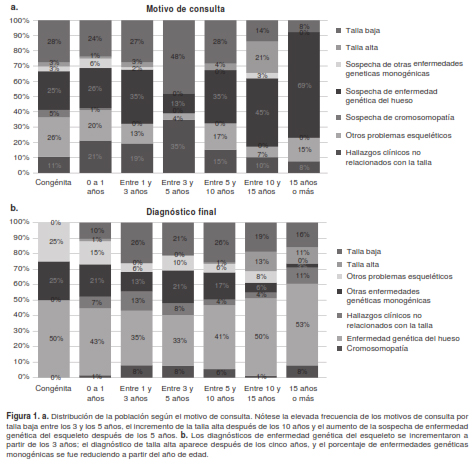
Los motivos de consulta más frecuentes en esta población fueron la sospecha de una enfermedad genética del esqueleto (30 %), la talla baja (26 %) y otros problemas esqueléticos (19 %) (cuadro 2).
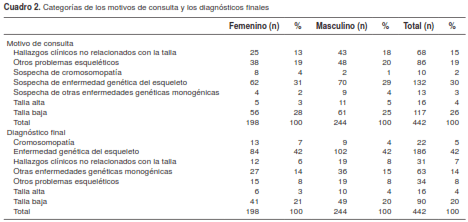
Al revisar las categorías de diagnóstico confirmado y sospecha, se encontró que en 42 % de la población se había registrado la enfermedad genética del esqueleto, seguida por la talla baja en el 20 % y otras enfermedades genéticas monogénicas en el 14 % (cuadro 2 y figura 3).
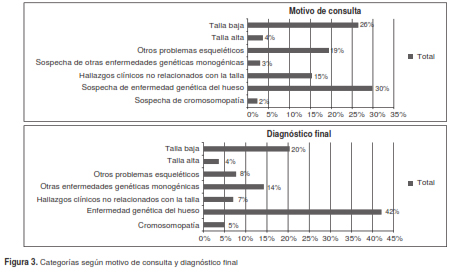
Los cinco diagnósticos finales más frecuentes sin categorización fueron: talla baja posnatal simétrica (53 pacientes), neurofibromatosis (43 pacientes), talla baja prenatal simétrica (29 pacientes), síndrome de Marfán (19 pacientes) y síndrome de Noonan (13 pacientes); muchos otros diagnósticos correspondieron a un solo caso (por ejemplo, el síndrome de Holt Oram o el síndrome de Smith Lemli Opitz), o se presentaron con una frecuencia menor de 3 %.
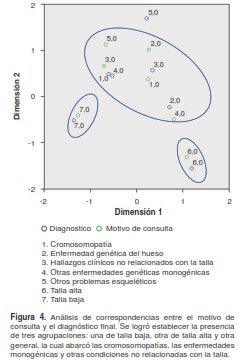
Al comparar los ítems de motivo de consulta y diagnóstico final, no se encontraron diferencias significativas entre ellos (p=1,94318). En el análisis de correspondencias de estas variables, no se establecieron conglomerados, lo cual ratificó la ausencia de diferencia en los datos mencionados, aunque sí se observó una preferencia por las variables de talla alta (6) y talla baja (7); además, estas mostraron una relación cercana entre el motivo de consulta y el diagnóstico final, y se diferenciaban por su comportamiento entre ellas dos y con las otras categorías (figura 4).
En 27,1 % de los 442 pacientes con sospecha de estas enfermedades se solicitaron estudios diagnósticos específicos, con resultados confirmatorios en 16,7 % de ellos. El 95 % de los pacientes evaluados no tenía antecedentes familiares de enfermedades relevantes; en cuanto al mecanismo de herencia de las enfermedades confirmadas y sospechadas, el 53,7 % de los casos se catalogaron como idiopáticos; el 32 %, como probablemente autosómicos dominantes; el 7,9 % como autosómicos recesivos; en el 5,4 % no aplicaba, y el 0,9 %, como ligados al cromosoma X.
En cuanto a los tipos de tratamiento, se consideraron las opciones de ausencia de tratamiento, tratamiento con un único medicamento o con varios, así como las intervenciones quirúrgicas, de soporte y la ‘ortesis’. El porcentaje acumulado de pacientes con algún tipo de soporte (terapias física, ocupacional o del lenguaje) fue de 36,6 %, con intervenciones quirúrgicas (22,8 %), con manejo farmacológico (19,9 %) y con ‘ortesis’ (9,9 %), en tanto que el 40,9 % no recibía ningún tratamiento.
El 14,7 % de los pacientes había sido sometido a algún tipo de cirugía ortopédica y al menos el 5 % requirió mas de una intervención quirúrgica. Según la categoría de diagnóstico (confirmado o sospecha), las enfermedades genéticas del esqueleto fueron las que más frecuentemente se sometieron a intervenciones ortopédicas, seguidas por otras enfermedades monogénicas y otros problemas esqueléticos. En el grupo con talla alta, no se habían practicado intervenciones quirúrgicas.
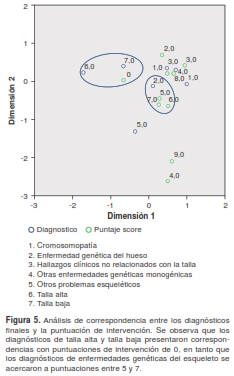
La ponderación de la puntuación de las intervenciones según el tipo de tratamiento se hizo a partir de un valor de 0 (ningún tratamiento) hasta 9 (tratamiento farmacológico, quirúrgico, uso de prótesis y soporte); dicha información se cruzó con el dato sobre el diagnóstico final (confirmado o sospecha) en otro análisis de correspondencias, en el cual se encontraron dos tendencias: una de puntajes de 5 a 7 en pacientes con diagnóstico de enfermedad genética del esqueleto, y otra de puntajes cercanos a 0 en aquellos con diagnósticos de talla alta o talla baja (figura 5).
Discusión
Durante el periodo de seis años, se atendieron 3.200 pacientes, aproximadamente, con impresión diagnóstica o sospecha de talla baja o enfermedades genéticas del esqueleto; en 11,7 % de ellos se solicitó valoración genética, cifra semejante a la encontrada en las remisiones de pacientes con talla baja hacia especialidades como la endocrinología o la gastroenterología pediátricas (15). Este motivo de consulta es uno de los más frecuentes en los servicios de genética (8,16), dada la importancia de establecer el diagnóstico etiológico en los niños, el cual frecuentemente es genético (enfermedades del esqueleto, tallas bajas idiopáticas originadas en el gen SHOX o síndromes monogénicos relacionados con la talla baja), así como el pronóstico clínico y el riesgo de recurrencia (17,18).
La distribución por sexo en la población de estudio fue semejante a la reportada en otras investigaciones como el estudio prospectivo de Flechter, et al. en 1.402 hombres (56,2 %) remitidos a un centro pediátrico de referencia entre 2004 y 2009 (19), y el de Majcher, et al., en 274 niños y 178 niñas con diagnóstico de talla baja, entre 1998 y 2009 (20). Esta tendencia ya ha sido objeto de análisis y se ha encontrado un menor número de remisiones a especialistas en la población femenina, lo que resulta en la ausencia de diagnóstico y en la falta de detección o el diagnóstico incorrecto o tardío de la condición, sobre todo cuando se trata de talla baja y se debe descartar el síndrome de Turner y el déficit de la hormona de crecimiento (15).
En cerca de la mitad de la población analizada (42,7 %), se reportaron signos congénitos con probable inicio de la enfermedad en el periodo prenatal tardío, probablemente relacionados con antecedentes como la condición de pequeño para la edad de gestación o con la restricción del crecimiento intrauterino. Sin embargo, al revisar el registro de síntomas prenatales, solo el 15,3 % de los pacientes lo presentaba, porcentaje que, probablemente, refleja solamente una parte del grupo afectado por enfermedades genéticas del esqueleto o cromosomopatías, pero no así el de talla baja de origen endocrino; a ello se añade que, en Colombia, la detección de displasias esqueléticas por ecografía obstétrica es muy baja (21). Estos valores contrastan con los reportados en otros estudios de prevalencia de enfermedades genéticas del esqueleto, como el de la población de Utah en 2012, con 55,6 % de casos de sospecha prenatal, de los cuales el 88 % resultó en aborto, lo que demuestra la necesidad de mejorar la sospecha clínica en la población local mediante los estudios prenatales de imágenes, para su posterior confirmación molecular y manejo integral y oportuno (22).
Se encontraron diferencias significativas entre la edad de inicio de los síntomas (2,58 años, en promedio) y la edad de diagnóstico (6,3 años, en promedio); llamó mucho la atención que la demora diagnóstica fue de casi cuatro años. Probablemente, varios factores influyeron en estas diferencias: la heterogeneidad de los diagnósticos de la población de estudio (176 códigos diagnósticos de CIE-10 y
195 diagnósticos finales, de los cuales los cinco primeros solo representaban el 31,3 %); el amplio espectro de edades de diagnóstico (figura 2) y la particularidad de que la detección de enfermedades genéticas del esqueleto en los pacientes se hizo en diversas edades (figura 1). Además, deben señalarse otros factores que podrían propiciar las diferencias entre la edad de inicio de síntomas y la del diagnóstico, tales como los relacionados con la calidad de la atención, la experiencia de los grupos encargados de la atención en el sistema de salud, la oportunidad en la atención, las barreras de acceso al sistema, el adecuado empleo de tecnologías de la salud y la falta de empoderamiento familiar frente a la condición de salud del menor, entre otros, los cuales podrían afectar, asimismo, las condiciones clínicas, sociales, económicas y emocionales de estos pacientes (23).
En este estudio, 75 % de los pacientes consultó por sospecha de enfermedad genética del esqueleto, talla baja y otros problemas esqueléticos, la cual se confirmó en el diagnóstico final como enfermedad genética del esqueleto en 42 % de los evaluados: como talla baja (20 %) relacionada con problemas endocrinológicos en 53 de aquellos con talla baja posnatal simétrica y en 29 de aquellos con talla baja prenatal simétrica, y como otras enfermedades genéticas monogénicas (14 %) en 13 casos de síndrome de Noonan y cuatro de síndrome de Russell-Silver, así como en otros con problemas esqueléticos (8 %). Al revisar otros estudios publicados, los datos de este contrastaron con los reportados por Flechtner, et al., en 2.546 pacientes con talla baja evaluados a lo largo de cinco años, 12 % de los cuales presentaba trastornos endocrinos, 8,9 %, trastornos de etiología genética, 5,1 %, condiciones crónicas, y 5,9 %, displasias óseas (19). En el centro de referencia del presente estudio, aparentemente se remite con mayor frecuencia a valoración genética a pacientes con diagnóstico de enfermedad genética de esqueleto, talla baja, otras enfermedades genéticas monogénicas u otros problemas esqueléticos, pues la frecuencia de defectos congénitos fue mayor en estos individuos. Es probable que en las clínicas de endocrinología pediátrica, se atienda un número más significativo de pacientes con diagnóstico de talla baja relacionada con el retardo constitucional del desarrollo, malnutrición, enfermedad celiaca, déficit de la hormona de crecimiento o talla baja familiar, que con enfermedades genéticas del esqueleto (24,25). Los diagnósticos finales con etiología clara de enfermedades genéticas del esqueleto correspondieron a 37,1 % (164 pacientes), distribuidos en 22 de los 40 grupos de la nosología y la clasificación, siendo el más frecuente el 29 (46 pacientes con enfermedades del grupo de componentes esqueléticos con desarrollo desorganizado), seguido por el 30 (36 pacientes con síndromes de crecimiento excesivo y compromiso esquelético), el 27 (17 pacientes con enfermedades de depósito lisosómico y compromiso esquelético), el 25 (11 pacientes con osteogénesis imperfecta u otras enfermedades con disminución de la densidad ósea), y el 4 (10 pacientes con alteraciones de la sulfatación), entre otros, lo cual contrasta ampliamente con el estudio de Flechtner, et al., en el cual se evaluaron pacientes con talla baja idiopática y pequeños para la edad de gestación, entre quienes los grupos preponderantes fueron el 1 (condrodisplasia, FGFR3) y el 17 (displasia mesomélica y rizomesomélica) (19). Es probable que estas diferencias se deban a que, en este estudio, el rango de edad de los pacientes era mayor (0 a 21 años; media de edad: 9,5 años) y las posibilidades de acceso al diagnóstico confirmatorio mediante métodos moleculares también lo eran. El presente estudio se enfocó en la elección de los códigos diagnósticos de la CIE-10 relacionados con la talla baja y las enfermedades genéticas del esqueleto; posteriormente, se clasificó a los pacientes en siete grupos según los motivos de consulta y en otros siete según el diagnóstico final, dado que las clasificaciones endocrinológicas y genéticas generales eran muy heterogéneas y difíciles de extrapolar. A pesar de ello, en la población de estudio se pudieron diferenciar tres condiciones: “talla baja”, “talla alta” y “otros” (enfermedades que comprometen el esqueleto). Al evaluar el manejo de los pacientes, se encontró una gran diversidad de enfoques que incluía terapias, ‘ortesis’, tratamientos farmacológicos, intervenciones quirúrgicas y combinaciones de estos. Con base en la puntuación de las intervenciones diseñadas en el marco del estudio, se pudo establecer que los pacientes con enfermedades genéticas del esqueleto habían sido sometidos a una mayor cantidad de intervenciones terapéuticas, quizá debido a su conocida comorbilidad, seguidos de aquellos con cromosomopatías, en tanto que, en casos de talla alta o talla baja, poco se recurrió a la combinación de tratamientos. Ello respondería, primero, a que no se había comenzado el tratamiento por haberse hecho el primer diagnóstico en la institución del estudio; segundo, al desconocimiento de nuevos enfoques y terapias para enfermedades acompañadas de talla alta o baja, y tercero, a las dificultades de acceso al tratamiento especializado en el sistema de salud del país (consultas de genética o endocrinología pediátrica), entre otros. Dichos factores deben alertar a los profesionales de la salud sobre la necesidad de una mayor y más oportuna intervención en estos dos grupos de pacientes. Por último, es necesario hacer claridad sobre algunos aspectos metodológicos: 1) pudo haber algún sesgo de selección en la muestra, ya que se partió de un ejercicio académico de selección de los códigos de la CIE-10 para agrupar la gran mayoría de los diagnósticos relacionados con enfermedades genéticas del esqueleto; 2) pudo haber un factor de confusión en la categorización de los motivos de consulta y el diagnóstico definitivo, pues algunos pudieron estar superpuestos; 3) solamente el 16,7 % de los pacientes tenía diagnóstico confirmado, de modo que pudo haber errores de clasificación, ya que en la mayoría de ellos se apeló a elementos clínicos, bioquímicos y radiológicos, muchas veces no concluyentes, para diagnosticar estas enfermedades, y 4), dado que no se trataba de un estudio con seguimiento, la impresión diagnóstica pudo haber cambiado con el tiempo y, por ende, la categorización de los pacientes. A pesar de ello, este trabajo es uno de los primeros a nivel regional que ha descrito algunos elementos epidemiológicos, clínicos y de manejo de las enfermedades genéticas del esqueleto, lo cual contribuye a la adopción de decisiones sobre nuevos tipos de intervenciones y sobre el seguimiento de este tipo de pacientes.
A los genetistas, ortopedistas, endocrinólogos y pediatras, entre otros especialistas, encargados de la valoración de los pacientes en el Instituto de Ortopedia Infantil Roosevelt.
Los autores declaramos que no tenemos intereses económicos o personales relacionados con los resultados publicados en este artículo.
No hubo ninguna.
Correspondencia:Harvy Mauricio Velasco, Instituto de Genética, Universidad Nacional de Colombia, Bogotá, D.C., Colombia Teléfono: (571) 316 5000, extensiones 11631 y 11620 hmvelascop@unal.edu.co y hmvelascop@unal.edu.co
1. Wit JM, Clayton PE, Rogol D, Savage MO, Saenger PH, Cohen P. Idiopathic short stature: Definition, epidemiology, and diagnostic evaluation. Growth Horm IGF Res. 2008;18:89-110. http://dx.doi.org/10.1016/j.ghir.2007.11.004.
2. Zahnleiter D, Uebe S, Ekici AB, Hoyer J, Wiesener A, Wieczorek D, et al. Rare copy number variants are a common cause of short stature. PLoS Genet. 2013;9:1-11. http://dx.doi.org/10.1371/journal.pgen.1003365.
3. Strufaldi MWL, Silva EM, Puccini RF. Follow-up of children and adolescents with short stature: The importance of the growth rate. São Paulo Med J. 2005;123:128-33. http://dx.doi.org/10.1590/S1516-31802005000300008
4. Neufeld L, Rubio M, Pinzón L, Tolentino L. Nutrición en Colombia: estrategia de país 2011-2014. Banco Interamericano de Desarrollo. 2010. Fecha de consulta: 1° de mayo de 2015. Disponible en: http://idbdocs.iadb.org/wsdocs/getdocument.aspx?docnum=35791560.
5. Fonseca Z, Heredia A, Ocampo P, Forero Y, Sarmiento O, álvarez M, et al. Encuesta Nacional de Situación Nutricional en Colombia 2010. Fecha de consulta: 1° de mayo de 2015. Disponible en: http://www.icbf.gov.co/portal/page/portal/PortalICBF/Bienestar/ENSIN1/ENSIN2010/ LibroENSIN2010.pdf.
6. Cohen P, Rogol D, Deal CL, Saenger P, Reiter EO, Ross JL, et al. Consensus statement on the diagnosis and treatment of children with idiopathic short stature: A summary of the Growth Hormone Research Society, the Lawson Wilkins Pediatric Endocrine Society, and the European Society for Paediatric Endocrinology Workshop. J Clin Endocrinol Metab. 2008;93:4210-7. http://dx.doi.org/10.1210/jc.2008-0509
7. Wit JM, Kiess W, Mullis P. Genetic evaluation of short stature. Best Pract Res Clin Endocrinol Metab. 2011;25:1- 17. http://dx.doi.org/10.1016/j.beem.2010.06.007
8. Acosta J, Zarante I. Consulta de genética médica en un hospital de segundo nivel en Colombia: impacto médico y social. Revista Salud Bosque. 2011;1:25-32.
9. Pombo M, Castro L, Rodríguez PC. El niño de talla baja. Protoc Diagn Ter Pediatr. 2011:1:236-54.
10. Del Toro KY, Durán P, Llano M. Enfoque del paciente con talla baja en pediatría. CAAP. 2008;7:28-37. Fecha de consulta: 1° de mayo de 2015. Disponible en: https://scp.com.co/precop-old/precop_files/modulo_7_vin_2/28-37%20Enfoque.pdf
11. Wit JM, Ranke MB, Kelnar CJ. Editores. The ESPE classification of paediatric endocrine diagnoses. Basel: Editorial Karger; 2007. p. 129.
12. Hall CM. International nosology and classification of constitutional disorders of bone (2001). Am J Med Genet. 2002; 113:65-77. http://dx.doi.org/10.1002/ajmg.10828
13. Superti-Furga A, Unger S. Nosology and classification of genetic skeletal disorders: 2006 revision. Am J Med Genet A. 2007;143:1-18. http://dx.doi.org/10.1002/ajmg.a.31483
14. Warman ML, Cormier-Daire V, Hall C, Krakow D, Lachman R, LeMerrer M, et al. Nosology and classification of genetic skeletal disorders: 2010 revision. Am J Med Genet A. 2011;155:943-68. http://dx.doi.org/10.1002/ajmg.a.33909
15. Grimberg A, Feemster KA, Pati S, Ramos M, Grundmeier R, Cucchiara AJ, et al. Medically underserved girls receive less evaluation for short stature. Pediatrics. 2011;127:696-702. http://dx.doi.org/10.1542/peds.2010-1563.
16. Esmer C, Urraca N. Patient follow-up is a major problem at genetics clinics. Am J Med Genet A. 2004;125:162-6. http://dx.doi.org/10.1002/ajmg.a.20303
17. Seaver LH, Irons M, American College of Medical Genetics (ACMG) Professional Practice and Guidelines Committee. ACMG practice guideline: Genetic evaluation of short stature. Genet Med. 2009;11:465-70. http://dx.doi.org/10.1097/GIM.0b013e3181a7e8f8
18. Sewell MD, Chahal A, Al-Hadithy N, Blunn GW, Molloy S, Hashemi-Nejad A. Genetic skeletal dysplasias: A guide to diagnosis and management. J Back Musculoskelet Rehabil. 2015;28:575-90. http://dx.doi.org/10.3233/BMR-140558
19. Flechtner I, Lambot-Juhan K, Teissier R, Colmenares A, Baujat G, Beltrand J, et al. Unexpected high frequency of skeletal dysplasia in idiopathic short stature and small for gestational age patients. Eur J Endocrinol. 2014;170:677-84. http://dx.doi.org/10.1530/EJE-13-0864
20. Majcher A, Witkowska-Sędek E, Bielecka-Jasiocha J, Pyrzak B. Causes of short stature in children in relation to their midparental heigh. Medycyna Wieku Rozwojowego. 2012;16:89-95.
21. Gómez J, Fernández N, Páez P, Zarante I. Detección de anomalías congénitas en 12.760 nacimientos de tres hospitales en la Ciudad de Bogotá, Colombia, 2004-2005 mediante ecografía prenatal. Rev Colomb Obstet Ginecol. 2007;58;194-201.
22. Konstantinidou AE, Agrogiannis G, Sifakis S, Karantanas A, Harakoglou V, Kaminopetros P, et al. Genetic skeletal disorders of the fetus and infant: Pathologic and molecular findings in a series of 41 cases. Birth Defects Res A Clin Mol Teratol. 2009;85:811-21. http://dx.doi.org/10.1002/bdra.20617.
23. Arkader A. Multiple hereditary exostoses: Its burden on childhood and beyond: Commentary on an article by A.L. Goud, MD, et al.: “Pain, physical and social functioning, and quality of life in individuals with multiple hereditary exostoses in The Netherlands. A national cohort study”. J Bone Joint Surg Am. 2012;94:e81. http://dx.doi.org/10.2106/JBJS.L.00277
24. Zargar AH, Laway BA, Masoodi SR, Wani AI, Salahuddin M. An etiological profile of short stature in the Indian subcontinent. J Paediatr Child Health. 1998;34:571-6. http://dx.doi.org/10.1046/j.1440-1754.1998.00308.x
25. Sultan M, Afzal M, Qureshi SM, Aziz S, Lutfullah M, Khan SA, et al. Etiology of short stature in children. J Coll Physicians Surg Pak. 2008;18:493-7. http://dx.doi.org/08.2008/JCPSP.493497