ARTÍCULO ORIGINAL
doi: http://dx.doi.org/10.7705/biomedica.v36i0.2984
1 Grupo de Virología, Vicerrectoría de Investigaciones, Universidad El Bosque, Bogotá, D.C., Colombia
2 Grupo de Patogénesis Infecciosa, Facultad de Odontología, Universidad Nacional de Colombia, Bogotá, D.C., Colombia
Contribución de los autores:
Jaime E. Castellanos: ejecución del proyecto, análisis e interpretación de datos
José I. Neissa: aplicación de la metodología
Sigrid J. Camacho: análisis e interpretación de datos
Todos los autores participaron en la redacción del manuscrito.
Recibido: 28/07/15; aceptado: 14/04/16
Introducción. El dengue es una enfermedad humana producida por el virus del mismo nombre, que se transmite por la picadura de mosquitos del género Aedes . La infección tiene una amplia gama de presentaciones clínicas que van desde la ausencia de síntomas hasta los casos fatales y afecta principalmente a la población pediátrica. Según la nueva clasificación de la enfermedad, las manifestaciones neurológicas se consideran un criterio para el diagnóstico del dengue grave.
Objetivo. Evaluar los posibles mecanismos involucrados en la aparición de los signos neurológicos en una línea celular de neuronas humanas, como modelo de infección con el virus del dengue del serotipo 2 (DENV-2).
Materiales y métodos. Se evaluó la sensibilidad y la permisividad de la línea celular SH-SY5Y a la infección por el DENV-2; se encontró que la proporción entre infección y producción viral era similar a las de las células de primates usadas como control positivo de la infección.
Resultados. La infección indujo un efecto citopático en la línea celular de neuroblastoma caracterizado por un proceso de muerte apoptótica, con aumento en la proporción de células positivas al emplear los métodos de anexina V y TUNEL. Se encontró una regulación positiva del factor de necrosis tumoral alfa (TNF-α), y el tratamiento con un anticuerpo anti-TNF-α aumentó ligeramente la supervivencia de las células infectadas. La adición de TNF-α exógeno a los cultivos infectados potenció la muerte celular.
Conclusión. Estos resultados sugieren, en su conjunto, que la regulación positiva del TNF-α podría hacer parte del proceso que induce daño y muerte celular durante el desarrollo de la encefalitis por dengue.
Palabras clave: dengue, apoptosis, factor de necrosis tumoral alfa, supervivencia celular, manifestaciones neurológicas, encefalitis.
doi: http://dx.doi.org/10.7705/biomedica.v36i0.2984
Dengue virus induces apoptosis in SH-SY5Y human neuroblastoma cells
Introduction: Dengue is a human disease caused by a virus with the same name, which is transmitted by the bite of Aedes mosquitoes. The infection has a wide range of clinical presentations ranging from asymptomatic to fatal cases, with the pediatric population being the most susceptible. According to the new classification of the disease, the neurological manifestations are considered a criterion for the diagnosis of severe dengue.
Objective: To evaluate the possible mechanisms involved in the onset of neurological signs in a cell line of human neurons as a model of infection with dengue virus type 2 (DENV-2).
Materials and methods: Susceptibility and permissiveness of the SH-SY5Y line to infection by DENV-2 was analyzed, showing that the proportions of viral infection and production are similar to those of primate cells used as positive control for infection.
Results: Infection induced a cytopathic effect on the neuroblastoma line characterized by apoptotic cell death process, increasing the proportion of annexin V and TUNEL positive cells and an upregulation of TNF- α . Treatment with anti-TNF- α antibody increased slightly cell survival of infected cells. The addition of exogenous TNF- α to the infected cultures enhanced cell death.
Conclusion: These results as a whole suggest that the upregulation of TNF- α could be part of the process that induces cell damage and death in cases of dengue encephalitis.
Key words: Dengue, apoptosis, tumor necrosis factor-alpha, cell survival, neurologic manifestations, encephalitis.
doi: http://dx.doi.org/10.7705/biomedica.v36i0.2984
El virus del dengue (DENV) es un flavivirus que se transmite por la picadura de mosquitos hembra de la especie Aedes aegypti (1). La infección causa una enfermedad febril de resolución espontánea que en algunas ocasiones se presenta como un cuadro grave que puede conducir a la muerte (2). Colombia es uno de los países de Latinoamérica con mayores tasas anuales de dengue debido a que en la mayor parte del territorio nacional circulan tanto el vector como los cuatro serotipos del virus (DENV 1 al 4) (3).
Previamente, la enfermedad por dengue se clasificaba como dengue clásico o dengue hemorrágico, este último con signos graves de pérdida plasmática y choque hipovolémico. Con base en la experiencia de diversos centros de atención clínica en el mundo, la Organización Mundial de la Salud (OMS) planteó una nueva clasificación de la enfermedad que incluye criterios adicionales para el diagnóstico (4). El amplio espectro de manifestaciones clínicas se agrupó en dos categorías: dengue (con signos de alarma y sin ellos) y dengue grave, el cual se caracteriza por el aumento de la permeabilidad vascular, extravasación de plasma, hemorragias graves y la alteración de la función de órganos como el hígado, el corazón o el cerebro (4). En este último caso, condiciones como la hepatitis, la miocarditis o los síntomas neurológicos, se convierten en los signos centrales para el diagnóstico, más allá de las hemorragias (5,6)
Desde hace más de una década, los pediatras colombianos venían observando que alrededor de 8 % de los pacientes con dengue grave presentaban alteraciones neurológicas durante la infección o después de ella (7). Esta cifra es similar a la que recientemente se reportó en 7,4 % de los pacientes de un estudio en India (8). Hay gran controversia acerca de si los signos neurológicos asociados a la infección por el DENV se deben a infección directa del tejido nervioso o si son el resultado de la disfunción nerviosa asociada al daño o la falla en sistemas diferentes al neurológico (como ocurre, por ejemplo, en la encefalopatía hepática), o si se deben a la presencia y la circulación constante de mediadores inflamatorios sistémicos o al aumento de los niveles de metabolitos durante la infección, lo cual modula la función neurológica (9). En un estudio recientemente publicado, se confirmó la etiología viral en 93 de los 291 pacientes mayores de 15 años con diagnóstico de infección del sistema nervioso central, y el virus de la encefalitis japonesa fue el más frecuente (36 casos), en tanto que el segundo virus más frecuentemente detectado en el líquido cefalorraquídeo fue el DENV (19 casos); en 80 % de estos, la encefalitis fue el único signo orientador del diagnóstico de dengue grave (10).
Los signos neurológicos que se han reportado incluyen desde cefaleas intensas y vómito en niños, hasta alteraciones de la conciencia o la conducta, convulsiones, polineuropatías, síndrome de Guillain-Barré y mielitis transversa (11); en varios casos se ha podido encontrar ARN viral o anticuerpos en el líquido cefalorraquídeo, lo cual demuestra replicación en el sistema nervioso (12).
Se ha propuesto que la apoptosis y la regulación excesiva de citocinas estarían implicadas en el daño neuronal por la infección con dengue (13,14). Sin embargo, la relación entre estos dos procesos no ha sido aclarada aún y no se sabe cómo las citocinas podrían estar involucradas en el daño neurológico. Entre las citocinas que se han visto reguladas de forma exacerbada en el cerebro de ratones como reacción a la infección por el virus del dengue, se encuentra el factor de necrosis tumoral alfa (TNF-α), además del interferón (IFN) alfa y la interleucina 1 alfa (IL-1a) (14), lo cual plantea la posibilidad de que la regulación excesiva del TNF-α y el efecto consecuente sean parte del daño secundario infligido por el virus.
En estudios previos hemos descrito la sensibilidad de la línea celular de neuroblastoma SH-SY5Y a la infección por dengue (15), por lo cual en el presente trabajo se la utilizó en un modelo de infección neuronal por DENV del serotipo 2, el cual se reporta frecuentemente asociado a condiciones neurológicas (8,12,16) y, además, históricamente es el más frecuente en las Américas y en Colombia (17), con el propósito de evaluar la muerte celular como reacción a la infección.
Materiales y métodos
Células y cultivo celular
Se utilizaron las células comerciales SH-SY5Y (Cat. CRL-2266, ATCC), que son fenotípicamente neuronales y se han aislado a partir de la metástasis ósea de un neuroblastoma humano. Esta sublínea celular proviene de las células SK-N-SH y SK-SH5Y, ambas reportadas previamente como
sensibles a la infección por dengue (18,19). Los experimentos se realizaron durante los pasajes 6 a 15. En algunos experimentos se utilizaron como controles positivos las células LLC-MK2 de riñón de Macaca mulatta (mono Rhesus ), de morfología epitelial, ampliamente reconocidas como sensibles a la infección por el virus del dengue. Todas las células se mantuvieron en medio DMEM con suero fetal bovino (SFB) al 10 % y antibióticos (100 U/ml de penicilina y 100 µg/ml de estreptomicina), en un ambiente de CO 2 al 5 % y una temperatura de 37 °C. Se confirmó que todos los cultivos celulares estuvieran libres de Mycoplasma spp. y se evaluaron diariamente mediante observación en el microscopio invertido.
Virus, infección y cuantificación del factor de necrosis tumoral alfa
Las infecciones se hicieron con DENV-2 proveniente del aislamiento clínico de un paciente (COL-789, pasaje 8), donado por el Grupo de Virología del Instituto Nacional de Salud de Colombia, el cual se cosechó en células C6/36 de Aedes albopictus en nuestro laboratorio y se confirmó por secuenciación.
La titulación viral (1,2 x 10 5 UFP/ml) se calculó evaluando la formación de placas de lisis en células LLC-MK2 de diluciones seriadas del inóculo. Con base en dicha titulación, se infectaron las células SH-SY5Y durante una hora con una multiplicidad de infección de 1, 0,5 o 0,1 (MOI: 1, MOI: 0,5 o MOI: 0,1); posteriormente, se retiró el inóculo y se permitió la infección durante uno a cuatro días. Como control se simuló una infección utilizando lisados de células C6/36 no infectados y, en algunos experimentos, con DENV-2 inactivados por rayos ultravioleta durante una hora.
Los cultivos infectados y los controles se evaluaron diariamente en el microscopio invertido para detectar la presencia del efecto citopático o de cambios relacionados con la muerte celular, tales como desprendimiento, pérdida de volumen, aumento de la refracción, disminución de la densidad del cultivo o aparición de morfología anormal.
Las concentraciones del TNF-α en los sobrenadantes de los cultivos se cuantificaron mediante el sistema de ELISA DuoSet (Cat. DY210 R&D Systems), el cual usa un anticuerpo de captura; tras añadir las muestras, se agregó el anticuerpo de detección biotinilado más peroxidasa acoplada a estreptavidina. La concentración se halló haciendo la extrapolación en una curva estándar de concentraciones conocidas.
Inmunofluorescencia e inmunocitoquímica indirectas
Se evaluó la presencia de antígeno viral en el interior de las células SH-SY5Y mediante técnicas de inmunocitoquímica e inmunoflurescencia indirectas y utilizando un anticuerpo monoclonal antiflavivirus (Cat. MAB8744, Chemicon), el cual reconoce eficientemente la proteína de envoltura de todos los flavivirus (20) y ha demostrado su utilidad tanto en células como en tejidos (21). Como control negativo se emplearon células SH-SY5Y inoculadas para simular la infección y, como control positivo, células LLC-MK2, cuya sensibilidad frente a la infección por el virus del dengue es ampliamente reconocida.
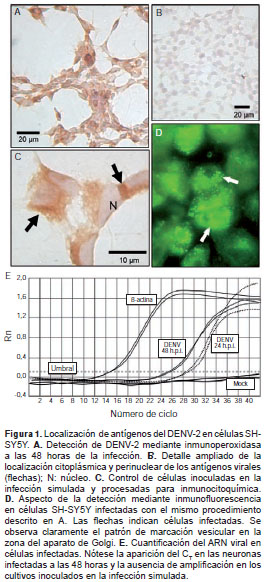
Se implementó un protocolo de inmunoperoxidasa en el cual las células se sembraron sobre laminillas de vidrio tratadas previamente con EHS Cell Attachment Matrix (Cat. G5971, Promega); a las 48 horas de infección se fijaron con paraformaldehído al 4 % durante 30 minutos y a continuación se les aplicó una protección con gelatina al 1 %. Se hizo la permeabilización y la inactivación de peroxidasas endógenas y, posteriormente, se bloquearon los sitios inespecíficos con suero de bovino recién nacido al 10 % en solución tampón fosfato salina (PBS), se adicionó el anticuerpo primario anti-flavivirus y, después de lavar con PBS, se agregó un anticuerpo secundario biotinilado producido en cabra (Cat. BA-9200, Vector); por último, se agregó estreptavidina acoplada a peroxidasa (Cat. SA-5004, Vector) durante 30 minutos y se hizo un revelado usando una mezcla en una proporción de 1:1 de H 2 O 2 al 0,02 % y diaminobenzidina en solución tampón Tris-HCl.
Después de colorear y deshidratar las células, se montaron en láminas para su observación en el microscopio y se fotografiaron con una cámara digital Nikon Coolpix 995. Para obtener los porcentajes de infección, se fotografiaron ocho campos en tres laminillas diferentes de tres réplicas independientes, y se contaron las células totales y aquellas que estaban marcadas para el antígeno del DENV (n=9).
El protocolo utilizado durante la inmunofluorescencia fue similar: el anticuerpo secundario en este caso fue un anti-IgG de ratón acoplado a isotiocianato de fluoresceína (FITC) (Cat. 74641, Sanofi), con dilución de 1/20 en suero de bloqueo (de bovino recién nacido) al 5 % en PBS, y se incubó durante 30 minutos a temperatura ambiente. Se hicieron dos lavados con PBS y uno con agua
destilada antes de hacer el montaje con glicerol. Las muestras se observaron y se fotografiaron en un microscopio de fluorescencia (Laborlux, Leitz).
Formación indirecta de placas
Para evaluar la adsorción y la penetración del DENV en las células de neuroblastoma (sensibilidad), se utilizó una técnica reportada previamente en hepatocitos por Chingsuwanrote, et al. (22), la cual permite obtener un dato confiable y linear en células que no forman monocapa. Las células SH-SY5Y se inocularon con una MOI:1 a 37 °C para permitir la adhesión y la penetración del virus durante dos horas, al cabo de las cuales se retiró el inóculo y se aplicó un tratamiento con glicina ácida (pH 3,0) durante un minuto para inactivar los virus que no lograron penetrar la célula.
Después de la infección, las células se desprendieron con tripsina al 0,2 % en PBS, se contaron en una cámara de Neubauer y se hicieron diluciones que luego se sembraron sobre una monocapa confluente de células LLC-MK2; se dejaron en adhesión durante cuatro horas para después retirar el sobrenadante y agregar un medio de recubrimiento que evita la dispersión del virus y permite, por ello, la formación de placas de lisis. Este medio contenía suero fetal bovino al 6,2 %, el medio DMEM, bicarbonato al 7,5 % y carboximetilcelulosa al 3 %. La formación de placas se evaluó siete días después mediante tinción de las monocapas con cristal violeta.
Reacción en cadena de la polimerasa cuantitativa
Se extrajo ARN total utilizando el reactivo Trizol ® (Gibco) a partir de células infectadas y no infectadas, siguiendo las instrucciones del fabricante. El ARN purificado se trató con la endonucleasa DNasa I (Cat. 18047-019, Invitrogen) en una solución tampón de digestión. Luego se hizo una nueva purificación del ARN con una mezcla de fenol/cloroformo/isoamil-alcohol. El ARN obtenido se cuantificó en un espectrofotómetro a 260 nm.
A partir de 500 ng de ARN se hizo una transcripción inversa utilizando cebadores aleatorios (25 µg/µl) y la enzima M-MLV (200 U/ml) (Cat. M170A, Promega) a 42 °C durante una hora. A partir de este ADN complementario, se hizo la amplificación mediante reacción en cadena de la polimerasa (PCR) en tiempo real en presencia de la molécula SYBR Green (kit Dynamo HS, Finnzymes) y utilizando el equipo GeneAmp 5700 Sequence Detection System (Perkin-Elmer).
La reacción tuvo un volumen final de 20 µl que contenían 4 µl de ADN complementario de plantilla, 10 µl de la Master Mix 2X (ADN polimerasa de Thermus brockianus , SYBR Green I, solución tampón de PCR optimizada, 5 mM de MgCl 2 y desoxirribonucleótidos trifosfato ( dNTP ) incluidas las desoxiuridinas trifosfato (dUTP), 0,4 µl de colorante ROX y 0,1 µM de cebadores de sentido y antisentido.
La especificidad del producto amplificado se corroboró mediante el análisis de la curva de disociación del producto. Se hizo una cuantificación relativa después de normalizar las muestras con la expresión del gen de β-actina utilizando el método de 2 -??Ct , según el protocolo establecido previamente en nuestro laboratorio (23).
Los cebadores específicos fueron los siguientes: para el gen β-actina, B-act F 5´-GAT CAT TGC TCC TCC TGA GC y B-act R 5´-ACT CCT GCT TGC TGA TCC AC; para la detección del ARN del DENV-2, DV2F 5´-TCA ATA TGC TGA AAC GCG CGA GAA ACC G y DV2R 5´-CGC CAC AAG GGC CAT GAA CAG (24); para la cuantificación de los mensajeros de citocinas, IL1bF 5´-TTG AAA GAT GAT AAG CCC ACT CTA y IL1bR 5´-CAG ACT CAA ATT CCA GCT TGT TA; para IL-6 se usaron IL6F 5´-GAC AGC CAC TCA CCT CTT CA y IL6R 5´-CCT CTT TGC TGC TTT CAC AC, y para el TNF-α, los cebadores TNFR 5´-CCC TGG TAT GAG CCC ATC TAT y TNFF 5´-AGG GCA ATG ATC CCA AAG TA.
Prueba de viabilidad celular
Para cuantificar la viabilidad celular se llevó a cabo una prueba de MTT (bromuro de 3-(4,5-dimetil-tiazol-2-il)-2,5-difeniltetrazolio, cat. M5655, Sigma), el cual es originalmente de color amarillo y, cuando se reduce a formazán, toma un color violeta mediante una reacción enzimática por la acción de deshidrogenasas celulares, lo cual permite demostrar la integridad metabólica.
Las células se sembraron en cajas de 96 pozos y se inocularon con DENV-2 o con el control; a continuación, se incubaron por 24, 48, 72 y 96 horas, y para evaluar la viabilidad, se les añadió MTT disuelto en DMEM fresco en una concentración final de 1 mg/ml, y se las incubó a 37 °C durante dos horas. Después de la corroboración microscópica de la formación de cristales de formazán, estos se disolvieron con la adición de 100 µl de dimetilsulfóxido (DMSO) y se cuantificó la absorbancia de cada pozo en 570 nm.
Método TUNEL
Se utilizó el método TUNEL ( TdT-mediated dUTP-biotin nick end labeling ) para detectar la fragmentación del ADN, la cual es signo de apoptosis, utilizando para su posterior detección la enzima desoxinucleotidil transferasa terminal (TdT), la cual marca dichos fragmentos incorporando dUTP biotinilados en los extremos 3´.
Las células se sembraron en laminillas de vidrio tratadas previamente con poli-L-lisina y, después de 48 horas, se fijaron y permeabilizaron para incubarlas con la enzima TdT recombinante (Cat. 3333574, Roche) en una concentración de 0,5 U/ml y en presencia de 0,2 nM de dUTP biotinilado en 2,5 mM de solución tampón de reacción y CoCl 2 , según lo recomendado por el fabricante. Se añadió, finalmente, estreptavidina acoplada al fluorocromo CY3 y, después de lavar, se hizo un montaje húmedo para visualizar y contar las células en el microscopio de fluorescencia.
Como control positivo se utilizaron células permeabilizadas y tratadas con 2 µg/ml de DNasa I durante 15 minutos a 37 °C. Como control negativo se utilizaron cultivos de células SH-SY5Y no infectados. Los núcleos positivos se contaron en, al menos, cuatro campos diferentes hasta completar un mínimo de 500 células en tres experimentos independientes, y los datos se expresaron como el porcentaje de células positivas por el método TUNEL.
Ensayo de fragmentación del ADN
Se extrajo el ADN de las células SH-SY5Y y se separó en un gel de agarosa para determinar la presencia del patrón en escalera característico de la apoptosis ( DNA laddering ), utilizando un procedimiento previamente descrito (25). Para ello, se sembraron 50.000 células por pozo en cajas de 12 pozos y 48 horas después de infectadas con DENV-2 se recolectaron y se les agregó 20 µl de solución tampón de lisis (2 mM de EDTA/100mM Tris-Cl, pH 8,0/0,8 % de SDS) y, a continuación, 2 µl de ribonucleasa A (50 mg/ml), mezcla que se incubó durante 30 minutos a 37 °C. Posteriormente, se añadieron 10 µl de proteinasa K (20 mg/ml) y se incubó durante dos horas a 50 °C, después de lo cual se agregaron 4 µl de solución tampón de carga azul de bromofenol 10X. Las muestras se separaron en una electroforesis en agarosa SeaKem al 1,8 % que contenía 0,6 µg/ml de bromuro de etidio, para su observación en un transiluminador UV.
Evaluación de la integridad de la membrana y de la externalización de la fosfatidilserina
Se evaluó la iniciación del proceso apoptótico y se lo cuantificó mediante el uso de anexina V acoplada a FITC; se cuantificaron, asimismo, las células necróticas mediante tinción con yoduro de propidio (Cat. Annex100F, AbD Serotec) en un citómetro de flujo, utilizando el protocolo recomendado por el fabricante.
En resumen, las células se desprendieron mediante tratamiento con tripsina al 0,2 % en PBS durante un minuto. Después de lavarlas, 10 6 células/ml se suspendieron de nuevo en 190 µl de solución tampón de unión, y se agregaron 4 µl de anexina V-FITC y 6 µl de yoduro de propidio, 20 µg/ml; a continuación, la muestra se incubó durante 10 minutos en la oscuridad a temperatura ambiente y se pasó por un citómetro de flujo (FACScan, Becton Dickinson). Como control positivo se utilizaron células SH-SY5Y expuestas a formalde-hído al 4 % en solución tampón de unión durante 30 minutos, según la indicación del fabricante. Como control negativo se utilizaron células SH-SY5Y sin ningún tratamiento y marcadas con anexina V e IP.
Ensayos de adición y neutralización del factor de necrosis tumoral alfa
En estos ensayos se utilizó el factor de necrosis tumoral alfa (TNF-α) recombinante humano expresado en levadura (Cat. T0157, Sigma). Se empleó el anticuerpo monoclonal neutralizador anti-TNF-α (R&D Systems. Cat. MAB610).
Se sembraron 1,4 x 10 5 células por pozo en cajas de 24 pozos; 24 horas después, se retiró el suero según un protocolo previamente descrito (26) y, al cabo de otras 24 horas, se hicieron infecciones con MOI de 1, 0,5 o 0,1; después de 90 minutos de adhesión y adsorción viral, se retiró el sobrenadante y se cambió por TNF-α recombinante humano en diferentes concentraciones disuelto en DMEM con SFB al 2,5 % y albúmina sérica bovina al 0,25 %.
En los ensayos de neutralización, el TNF-α recombinante y el anticuerpo anti-TNF-α se dejaron en incubación durante dos horas a temperatura ambiente antes de agregarlos al cultivo. Pasadas 3 y 24 horas, las células se desprendieron con tripsina al 0,2 % para su análisis mediante citometría de flujo usando la marcación con anexina V y yoduro de propidio, según el protocolo descrito anteriormente.
Análisis estadístico
Los porcentajes obtenidos con el método TUNEL se sometieron a una transformación angular y, previa aplicación del test de Levene, se hizo una ANOVA de una vía seguida del test post hoc T3 de Scheffe. Un valor de p menor de 0,05 se consideró estadísticamente significativo.
Resultados
Las células SH-SY5Y son sensibles a la infección por DENV-2.
En la evaluación mediante inmunoperoxidasa a las 48 horas de la infección se observó que más de 90 % de las neuronas eran positivas para el antígeno viral (figura 1 A), el cual se ubicó alrededor del núcleo (figura 1 C). En la figura 1 B se muestra el aspecto de las células inoculadas en la infección simulada y procesadas con inmunoperoxidasa. En la inmunofluorescencia se pudo ver la intensa marcación citoplásmica (figura 1 D), la cual reflejó la abundancia del antígeno. Se cuantificó el genoma viral mediante una PCR en tiempo real a partir del ARN obtenido de una monocapa de células SH-SY5Y infectadas. El resultado mostró mayor cantidad de ARN viral a las 48 que a las 24 horas de la infección (C T 25,3±1,4 Vs . 29,6±2,1) (figura 1 E).
Mediante un ensayo de formación indirecta de placas se evaluó si las neuronas SH-SY5Y permitían la replicación del DENV-2 liberando viriones infecciosos al medio. Debido a que estas células crecen formando cúmulos que fácilmente se desprenden y que no alcanzan una confluencia de 100 %, es imposible hacer una formación convencional de placas, por lo que se hizo una indirecta.
Las células SH-SY5Y inoculadas con DENV-2 liberaron viriones que formaron placas de lisis sobre las monocapas de LLC-MK2. El número de placas indirectas producidas por las células SH-SY5Y fue ligeramente mayor que el número de placas indirectas producidas por las células LLC-MK2 infectadas con igual MOI y aplicadas sobre monocapas de esta misma línea celular (figura 2 A y B). El porcentaje de células SH-SY5Y infectadas (2,5 %) fue superior al de las células LLC-MK2 (1,0 %) infectadas bajo las mismas condiciones (figura 2 C). El número de placas indirectas formadas por la inoculación de células SH-SY5Y sobre células LLC-MK2 aumentó significativamente a partir de las 12 horas de la infección (figura 2 D), lo cual indica que la liberación de la primera progenie del virus ocurrió entre las ocho y las 12 horas de la inoculación.
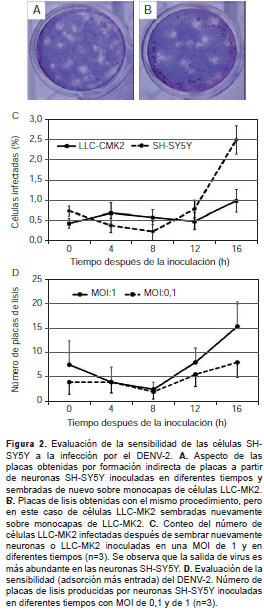
Este resultado confirmó que las células SH-SY5Y son sensibles a la infección por el DENV-2 y que constituyen un buen modelo de neuroinfección in vitro .
Las células SH-SY5Y disminuyen su viabilidad tras la infección con DENV-2.
Durante la infección se observaron cambios morfológicos indicativos del efecto citopático en las células SH-SY5Y. A las 72 horas de la infección, se observó pérdida de células adheridas, aumento del número de cúmulos, pérdida de la morfología poligonal y su reemplazo por una morfología redondeada, reducción en el número de neuritas, y citoplasmas con vesículas (figura 3 ).
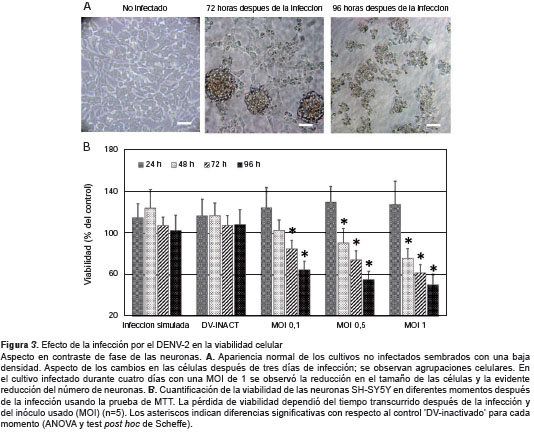
Todo lo anterior sugiere que la infección con DENV-2 inicia un proceso de muerte celular que culmina con la lisis o destrucción de la membrana. Se confirmaron estos hallazgos evaluando la viabilidad con la prueba de MTT. Como se muestra en la figura 3B, a las 96 horas, la infección con el DENV-2 indujo una importante disminución de la viabilidad celular de, aproximadamente, 50 % con una MOI de 1, de 45 % en una MOI de 0,5 y de 36 % en una MOI de 0,1, con respecto al control sin infectar. Curiosamente, durante las primeras 24 horas de infección se observó una tendencia al incremento en la absorbancia. No se observaron cambios en la medición de los cultivos inoculados en la infección simulada o con inóculos inactivados por radiación ultravioleta.
La infección con DENV-2 induce la expresión del TNF- a en las células SH-SY5Y.
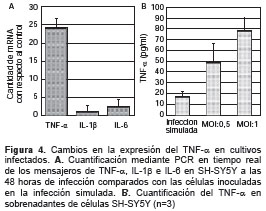
En las células SH-SY5Y infectadas con DENV-2 se encontró una regulación positiva en la transcripción y la traducción del TNF-α, en comparación con las células inoculadas en la infección simulada. A las 48 horas de la infección la regulación del mensajero fue 24 veces mayor en los cultivos infectados con el virus que en los infectados de forma simulada (figura 4A). No obstante, la transcripción de las citocinas inflamatorias IL-1β e IL-6 no se vio modificada de forma sustancial, lo cual indica que la expresión exacerbada del TNF-α es un fenómeno específico. A las 48 de la infección, la concentración del TNF-α en los cultivos infectados fue significativamente mayor que en los cultivos inoculados en la infección simulada. Los niveles de esta citocina fueron mayores en los cultivos infectados con una MOI de 1 que con una de 0,5 (figura 4B)
La infección con el DENV-2 induce apoptosis en las células SH-SY5Y.
Algunas de las características morfológicas observadas previamente podrían ser indicativas de la activación de un mecanismo apoptótico durante la infección de las células SH-SY5Y por el DENV-2 y, por esa razón, se evaluó la muerte neuronal apoptótica. En los cultivos con infección por el DENV-2 se incrementó sustancialmente el número de células positivas al aplicar el método TUNEL y el número en los cultivos inoculados con una MOI de 1 fue mayor que en aquellos con una de 0,1 (figura 5A).
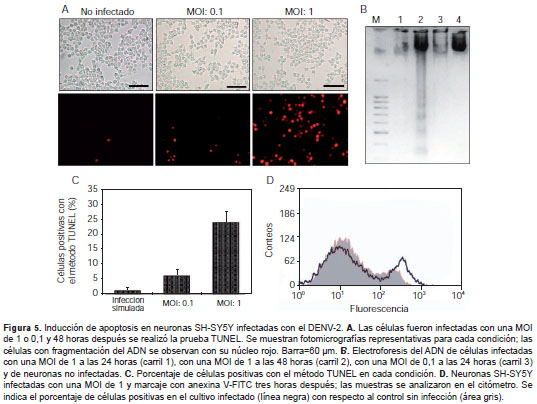
Los porcentajes de células positivas con el método TUNEL para el control inoculado en la infección simulada, y el de las inoculadas con una MOI de 0,1 y una de 1 fueron de 1,2 %, 6,1 % y 24,2 %, respectivamente (figura 5B). Aunque usualmente un resultado positivo al aplicar el método TUNEL se considera como prueba de un proceso apoptótico, estrictamente hablando la fragmentación del ADN puede encontrarse tanto en la apoptosis como en la necrosis, por lo cual, al evaluar el ADN purificado en estas células, se observó el distintivo patrón en escalera que caracteriza los procesos apoptóticos (figura 5C). Por el contrario, el ADN proveniente de células sin infectar permaneció intacto.
Por otro lado, para confirmar el proceso apoptótico en las células infectadas, se evaluó la translocación de la fosfatidilserina hacia la superficie externa como signo de apoptosis temprana. Las células SH-SY5Y se inocularon y, al cabo de tres horas de infección, fueron desprendidas y marcadas con anexina V, FITC y yoduro de propidio. Se detectó un aumento del número de células positivas con respecto a los cultivos sin infectar superior a 13 % en las infecciones con una MOI de 1 (figura 5D). El porcentaje de células positivas con el yoduro de propidio fue de 2,4 %, lo cual indica que en las etapas tempranas de la infección hay preponderancia del proceso apoptótico frente al necrótico. En su conjunto, estos experimentos demostraron que al menos parte del proceso de muerte celular inducido por el DENV-2 corresponde a un proceso apoptótico.
El TNF- a indujo un proceso de muerte apoptótica en las células SH-SY5Y.
Para evaluar el efecto del TNF-α en la viabilidad celular, las células SH-SY5Y se cultivaron y se estimularon con 200 ng/ml de esta citocina. Las células expuestas al TNF-α presentaron cambios morfológicos muy discretos a las 48 horas, pero claramente observables a las 72 horas de tratamiento, los cuales consistieron en aumento del número de células desprendidas, disminución del volumen celular, disminución de la densidad celular, aparición de una apariencia más redondeada y pérdida de la morfología poligonal, así como disminución del número y la longitud de las neuritas; además, algunas células también presentaron una morfología anormal con bordes irregulares.
Estos hallazgos indican muerte celular producida por apoptosis. Se encontró que, aproximadamente, 10 % de las células eran positivas con el método de anexina V después de 24 horas de tratamiento con el TNF-α en dosis de 50 y 200 ng/ml, y no se registraron cambios en los porcentajes de marcación con yoduro de propidio, lo cual indica que esta citocina indujo el proceso de muerte celular apoptótico en las células SH-SY5Y.
El TNF-αlfa incrementó la mortalidad celular inducida por el DENV-2.
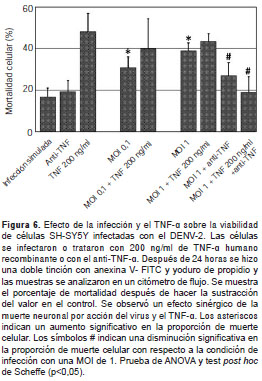
Los resultados presentados hasta este punto evidencian, en primer lugar, una regulación positiva en la expresión del TNF-α durante la infección por el DENV-2, además, un proceso de muerte en las células SH-SY5Y desencadenado por esta citocina, así como un proceso de muerte celular durante la infección por el DENV-2, eventos estos que podrían estar correlacionados. Para investigar la posible relación entre estos factores, se agregó la proteína TNF-α recombinante humana a cultivos de células SH-SY5Y infectadas con el DENV-2 y, paralelamente, se neutralizó su acción mediante un anticuerpo anti-TNF-α. Se consideró que había muerte celular en las neuronas que fueron positivas con el método de anexina-V, con yoduro de propidio o con ambos. Cuando se trataron las células con el TNF-α y simultáneamente se infectaron con el DENV-2, se observó que, a partir de las 24 horas, había una disminución de la viabilidad celular mayor que la producida con cualquiera de los dos factores por separado.
Resulta interesante que el tratamiento con el TNF-α y la infección con una MOI de 0,1 produjera una sinergia resultante en un aumento de la mortalidad superior, incluso, al producido por la infección con un inóculo 10 veces mayor (figura 6 ). El porcentaje de células muertas con respecto al control a las 24 horas de la infección aumentó casi dos veces con una MOI de 1 y disminuyó levemente después de la adición de un anticuerpo anti-TNF-α. La adición del TNF-α a los cultivos infectados con el DENV-2 incrementó significativamente la muerte celular, sin embargo, su neutralización evitó por completo el efecto sinérgico observado durante la infección con el DENV-2 y el tratamiento simultáneo con TNF-α. Esto sugiere que durante la infección de las células neuronales el TNF-α podría ser parte del proceso de muerte celular inducido por el DENV-2.
Discusión
Aunque el DENV no es un virus neurotrópico, hoy se considera la encefalitis por dengue como una verdadera entidad clínica y en algunas zonas endémicas los signos neurológicos pueden, incluso, llegar a ser la manifestación más frecuente del dengue grave (27,28). Durante mucho tiempo se consideró que las anormalidades neurológicas durante la infección por el DENV eran secundarias al daño hepático (encefalopatía), pero, desde los años 90 se ha acumulado evidencia sobre la presencia del virus o de anticuerpos específicos en el encéfalo y hasta el momento se han reportado más de 200 trabajos, principalmente series de casos, sobre las complicaciones neurológicas, sensoriales, neuromusculares y neurooftalmológicas en el curso del dengue (5). El aumento de la frecuencia de estos reportes sugiere, en primera instancia, que se han producido cambios en el perfil de la enfermedad debidos, probablemente, a variaciones en el tropismo del virus que promueven su interacción con diferentes tipos de moléculas y células receptoras. Ello resalta la necesidad de conocer más sobre el virus y sobre los posibles mecanismos a los que recurre para infectar diferentes tipos de células y tejidos (29,30).
Es poco lo que se sabe sobre el neurotropismo y la neuroinfección por el DENV, lo cual se debe, en parte, a que no hay un modelo animal que reproduzca los signos y síntomas característicos de la enfermedad, con lo cual se podrían conocer los posibles mecanismos asociados a la neuropatología (31). Para infectar ratones se requiere de la modificación de factores virales, del huésped o de ambos. Cuando esto se ha logrado, se ha detectado que las neuronas son el principal sitio de replicación viral en el sistema nervioso (30,32).
La investigación sobre los modelos in vitro continúa siendo de gran importancia y buena parte de la información obtenida sobre el dengue proviene de ellos. En el presente trabajo se demostró la sensibilidad y la permisividad de la línea neuronal humana SH-SY5Y a un DENV-2 aislado de muestras de pacientes, lo cual evidenció la transcripción del genoma y la traducción de las proteínas virales. Mediante un ensayo de formación indirecta de placas se confirmó, además, que las neuronas producen y liberan tempranamente viriones infecciosos; esta metodología no se había usado previamente y permitió, asimismo, evidenciar que la sensibilidad y la productividad de las células de neuroblastoma son similares a las de las células LLC-MK2, una de las líneas más sensibles al DENV (33).
En la mayoría de estudios con pacientes, las manifestaciones neurológicas se han observado más frecuentemente en aquellos infectados con el DENV-2 (8), un hallazgo que coincide con lo reportado por Araujo, et al . (11), y Solomon, et al . (12), en el sentido de que este serotipo posee características intrínsecas que propician su efecto en el sistema nervioso. En el presente trabajo, la infección con el DENV-2 causó un evidente efecto citopático en los cultivos de neuroblastoma, así como pérdida de viabilidad, la cual fue más evidente cuando se usó una mayor multiplicidad de infección. La evaluación morfológica y las pruebas TUNEL evidenciaron el proceso de muerte celular por apoptosis desde una fase muy temprana de la infección, lo cual se corroboró con el patrón de degradación del ADN y la marcación con anexina V, en concordancia con lo reportado en modelos de ratón con neuroinfección por dengue (13) y en otras líneas neuronales (34-38). Aunque anteriormente Lee, e t al. (39) reportaron que en células de neuroblastoma de ratón la infección con el DENV activaba un mecanismo anti-apoptótico dependiente de la vía PI3K/Akt, al parecer esto no sucede en las células SH-SY5Y. Las células HUVEC, que se infectan en más de 90 % (como ocurrió en este caso), inducen la apoptosis solo en cerca de 20 % de las células, al parecer como un mecanismo de potenciación de la replicación viral (40).
La infección con el DENV-2 indujo una regulación excesiva de los mensajeros de TNF-α, lo cual coincide con lo reportado previamente en cerebros de ratón (14), aunque la transcripción de otras citocinas inflamatorias, como IL-1β e IL-6, no aumentó. Es posible que estas dos últimas se produzcan en el cerebro por la acción de células diferentes a las neuronas. Se sabe muy bien que el TNF-α es el principal mediador inflamatorio durante la infección por el DENV (41,42) y que su concentración en el cerebro puede ser la suma del que producen las neuronas, más que resultado de su difusión desde la circulación sistémica hacia el parénquima cerebral.
Se ha demostrado que el TNF-α desempeña un papel dual y ambiguo de protección o daño neuronal dependiendo del contexto (43). El TNF-α exógeno indujo un aumento de la mortalidad de las células SH-SY5Y en un proceso definitivamente apoptótico (células positivas para anexina V), similar a lo reportado previamente (26,44). La inducción de la apoptosis dependiente del TNF-α suele asociarse a la unión de este ligando con el receptor TNFR 1 y a la consecuente activación de la vía extrínseca que involucra a la caspasa 8. Sin embargo, como parte de los mecanismos tumorales de evasión de la muerte celular, las células SH-SY5Y no expresan la caspasa 8, inactivación que se debe a un mecanismo epigenético dependiente de la metilación del promotor del gen (45,46). Como alternativa de activación de la vía extrínseca está la caspasa 10, cuya expresión normal se ha reportado en estas células (45) y merece evaluarse en el contexto de la infección por el DENV.
La adición del TNF-α en las células infectadas aumentó la mortalidad celular, actuando sinérgicamente con la infección por el DENV, en tanto que el uso de un anticuerpo anti-TNF-α disminuyó levemente la mortalidad causada por la infección, aunque sí abolió por completo el efecto sinérgico deletéreo de la infección bajo el estímulo del TNF-α. La proporción de la mortalidad en las neuronas infectadas no cambió con el empleo del anti-TNF-α. Esto sugiere que la participación del TNF-α autocrino en la mortalidad celular observada sería solo parcialmente responsable de la apoptosis, lo cual indicaría la participación de otros mecanismos inductores de muerte presentes durante la infección in vitro . En este sentido, se han reportado nueve aminoácidos del extremo C-terminal de la proteína M del DENV como necesarios y suficientes para la inducción de apoptosis en células de neuroblastoma en el ratón por la acción de un mecanismo dependiente de la vía mitocondrial (34). La ausencia de la caspasa 8 en el modelo indica una tendencia hacia un débil efecto apoptótico dependiente del TNF-α endógeno producido por los cultivos infectados, y hacia una mayor relevancia de otros mecanismos inductores de muerte que deben ser investigados.
Asimismo, se ha sugerido que, aunque la infección por dengue induce apoptosis en varios tipos celulares, también activa vías bioquímicamente diferentes de una manera específica para cada célula. Esta apreciación se fundamenta en que la expresión ectópica exacerbada del Bcl-2 retrasó el proceso apoptótico inducido por el DENV-2 en fibroblastos BHK-21, pero no en células de neuroblastoma de ratón N18. A su vez, a pesar de sus propiedades inductoras de apoptosis, la expresión de Bcl-Xs no produjo este efecto en las células BHK-21, aunque sí en las N18 (38). En este mismo sentido, Jan, et al. (36), reportaron que, en las células SK-N-SH, la infección con el DENV-2 activó vías relacionadas con la apoptosis, algunas de ellas no muy bien caracterizadas, y propusieron un modelo secuencial que involucra a la fosfolipasa A2, el ácido araquidónico, el anión superóxido y el complejo proteico NF-kB.
Más allá de la participación directa del virus en el proceso apoptótico neuronal, es posible que citocinas locales o sistémicas estén involucradas. Por ejemplo, se conoce que la infección cerebral con el DENV-2 en ratones indujo un incremento de mensajeros de TNF-α, y de IL1, IL6, IL1-β, entre otras (14). No hay información sobre estos aspectos en células humanas o sobre la probable participación de estas citocinas, en particular el TNF-α, en la muerte neuronal por dengue.
En su conjunto, los resultados del presente estudio sugieren que el TNF-α puede ser parte del proceso que induce daño y muerte celular durante una neuroinfección por dengue, aunque deberá evaluarse el efecto que pueda tener el TNF-α producido por otras células cerebrales, así como el efecto en la función cerebral de sus altas concentraciones en el plasma durante la infección aguda.
Aunque la proporción de casos con signos neurológicos durante la infección por el DENV es relativamente baja, se diagnostica cada vez con mayor frecuencia y abre un campo de investigación en torno a los cambios genéticos de los virus circulantes y a las características inmunitarias o genéticas de los huéspedes que podrían ser las responsables del daño nervioso que se presenta en esas ocasiones. En este estudio, se estableció que la muerte neuronal por apoptosis podría ser el mecanismo por el cual algunos aislamientos del DENV pueden tener un comportamiento virulento e invasivo en el sistema nervioso.
Los autores no tienen ningún conflicto de intereses con respecto a los productos o protocolos mencionados en este manuscrito.
Este trabajo fue financiado por la Vicerrectoría de Investigaciones de la Universidad El Bosque y por Colciencias (proyecto 1308-489-25267).
Correspondencia:
Jaime E. Castellanos, Grupo de Virología, Universidad El Bosque, Avenida Carrera 9 N° 131A-02, Bogotá, D.C., Colombia
Teléfono: (571) 648 9066 Fax: (571) 625 2030
castellanosjaime@unbosque.edu.co
1. Velandia ML, Castellanos JE. Virus del dengue: estructura y ciclo viral. Infectio. 2011;15:33-43. http://dx.doi.org/10.1016/S0123-9392(11)70074-1
2. Póvoa TF, Alves AM, Oliveira CA, Nuovo GJ, Chagas VL, Paes MV. The pathology of severe dengue in multiple organs of human fatal cases: Histopathology, ultrastructure and virus replication. PLoS One. 2014;9:e83386. http://dx.doi.org/10.1371/journal.pone.0083386
3. Villar L, Rojas D, Besada-Lombana S, Sarti E. Epidemiological trends of dengue disease in Colombia (2000-2011): A systematic review. PLoS Negl Trop Dis. 2015;9:e0003499. http://dx.doi.org/10.1371/journal.pntd.0003499
4. Organización Mundial de la Salud, Programa Especial para Investigación y Capacitación en Enfermedades Tropicales. Dengue. Guías para el diagnóstico, tratamiento, prevención y control. La Paz: OPS/OMS; 2010.
5. Castellanos J, Bello J, Velandia-Romero M. Manifestaciones neurológicas durante la infección por el virus del dengue. Infectio. 2014;18:167-76. http://dx.doi.org/10.1016/j.infect.2014.02.006
6. Nimmagadda SS, Mahabala C, Boloor A, Raghuram PM, Nayak UA. Atypical manifestations of dengue fever (DF) - Where do we stand today? J Clin Diagn Res. 2014;8:71-3. http://dx.doi.org/10.7860/JCDR/2014/6885.3960
7. Méndez A, González G. Manifestaciones clínicas inusuales del dengue hemorrágico en niños. Biomédica. 2006;26:61-70. http://dx.doi.org/10.7705/biomedica.v26i1.1395
8. Neeraja M, Iakshmi V, Teja VD, Lavanya V, Priyanka EN, Subhada K, et al . Unusual and rare manifestations of dengue during a dengue outbreak in a tertiary care hospital in South India. Arch Virol. 2014;159:1567-73. http://dx.doi.org/10.1007/s00705-014-2010-x
9. Jackson S, Hann-Chu J, Chia P, Morgan O, Ng L. Dengue encephalitis. En: Ruzek D, editor. Flavivirus encephalitis. Croatia: InTech; 2011. p. 71-88.
10. Tan LV, Thai LH, Phu NH, Nghia HD, Chuong LV, Sinh DX, et al. Viral etiology of central nervous system infections in adults admitted to a tertiary referral hospital in Southern Vietnam over 12 years. PLoS Negl Trop Dis. 2014;8:e3127. http://dx.doi.org/10.1371/journal.pntd.0003127
11. Araújo F, Nogueira R, Araujo MS, Perdigão A, Cavalcante L, Brilhante R, et al . Dengue in patients with central nervous system manifestations, Brazil. Emerg Infect Dis. 2012;18:677-9. http://dx.doi.org/10.3201/eid1804.111552
12. Solomon T, Dung NM, Vaughn DW, Kneen R, Thao LT, Raengsakulrach B, et al . Neurological manifestations of dengue infection. Lancet. 2000;355:1053-9. http://dx.doi.org/10.1016/S0140-6736(00)02036-5
13. Desprès P, Frenkiel M, Ceccaldi P, Dos Santos CD, Deubel V. Apoptosis in the mouse central nervous system in response to infection with mouse-neurovirulent dengue viruses. J Virol. 1998;72:823-9.
14. Sánchez-Burgos G, Hernández-Pando R, Campbell IL, Ramos-Castañeda J, Ramos C. Cytokine production in brain of mice experimentally infected with dengue virus. Neuroreport. 2004;15:37-42.
15. García-Rivera EJ, Rigau-Pérez JG. Encephalitis and dengue. Lancet. 2002;360:261. http://dx.doi.org/10.1016/S0140-6736(02)09481-3
16. Rincón V, Alvear D, Solano O, Prada-Arismendy J, Castellanos JE. La infección por virus dengue induce la disminución de marcadores de diferenciación en células de neuroblastoma. Iatreia. 2011;24:126-35.
17. Padilla J, Rojas D, Sáenz-Gómez R. Dengue en Colombia. Epidemiología de la reemergencia a la hiperendemia. Bogotá, D.C.: Guías de Impresión Ltda; 2012. p. 249.
18. Ramos-Castañeda J, Imbert JL, Barron BL, Ramos C. A 65-kDa trypsin-sensible membrane cell protein as a possible receptor for dengue virus in cultured neuroblastoma cells. J Neurovirol. 1997;3:435-40.
19. Reyes-Del Valle J, Chávez-Salinas S, Medina F, Del Angel RM. Heat shock protein 90 and heat shock protein 70 are components of dengue virus receptor complex in human cells. J Virol. 2005;79:4557-67. http://dx.doi.org/10.1128/JVI.79.8.4557-4567.2005
20. Huelseweh B, Ehricht R, Marschall HJ. A simple and rapid protein array based method for the simultaneous detection of biowarfare agents. Proteomics. 2006;6:2972-81. http://dx.doi.org/10.1002/pmic.200500721
21. Velandia-Romero ML, Acosta-Losada O, Castellanos JE. In vivo infection by a neuroinvasive neurovirulent dengue virus. J Neurovirol. 2012;18:374-87. http://dx.doi.org/10.1007/s13365-012-0117
22. Chingsuwanrote P, Suksanpaisan L, Smith DR. Adaptation of the plaque assay methodology for dengue virus infected HepG2 cells. J Virol Methods. 2004;116:119-21. http://dx.doi.org/10.1016/j.jviromet.2003.11.002
23. Prada-Arismendy J, Castellanos JE. Real time PCR. Application in dengue studies. Colomb Med. 2011;42:243-58.
24. Lanciotti RS, Calisher CH, Gubler DJ, Chang GJ, Vorndam AV. Rapid detection and typing of dengue viruses from clinical samples by using reverse transcriptase-polymerase chain reaction. J Clin Microbiol. 1992;30:545-51.
25. Zhivotovsky B, Orrenius S. Assessment of apoptosis and necrosis by DNA fragmentation and morphological criteria. Curr Protoc Cell Biol. 2001;18. http://dx.doi.org/10.1002/0471143030.cb1803s12
26. Kweon SM, Lee ZW, Yi SJ, Kim YM, Han JA, Paik SG, et al . Protective role of tissue transglutaminase in the cell death induced by TNF- α in SH-SY5Y neuroblastoma cells. J Biochem Mol Biol. 2004;37:185-91.
27. Solbrig M, Perng G. Current neurological observations and complications of dengue virus infection. Curr Neurol Neurosci Rep. 2015;15:29. http://dx.doi.org/10.1007/s11910- 015-0550-4
28. Carod-Artal FJ, Wichmann O, Farrar J, Gascón J. Neurological complications of dengue virus infection. Lancet Neurol. 2013;12:906-19. http://dx.doi.org/10.1016/S1474-4422(13)70150-9
29. Domínguez R, Kuster G, Onuki-Castro F, Souza V, Levi J, Pannuti C. Involvement of the central nervous system in patients with dengue virus infection. J Neurol Sci. 2008;267:36-40. http://dx.doi.org/10.1016/j.jns.2007.09.040
30. Velandia M, Castellanos JE. Flavivirus neurotropism, neuroinvasion, neurovirulence and neurosusceptibility: Clues to understanding flavivirus- and dengue-induced encephalitis. In: García ML, Romanowski V, editors. Viral genomes - molecular structure, diversity, gene expression mechanisms and host-virus interactions. Croacia: InTech; 2012. p. 219-40. http://dx.doi.org/10.5772/26571
31. Charlier N, Leyssen P, De Clercq E, Neyts J. Rodent models for the study of therapy against flavivirus infections. Antiviral Res. 2004;63:67-77. http://dx.doi.org/10.1016/j.antiviral.2004.06.007
32. Imbert J, Guevara P, Ramos-Castañeda J, Ramos C, Sotelo J. Dengue virus infects mouse cultured neurons but not astrocytes. J Med Virol. 1994;42:228-33.
33. Guzmán MG, Kouri G. Advances in dengue diagnosis. Clin Diagn Lab Immunol. 1996;3:621-7.
34. Catteau A, Kalinina O, Wagner MC, Deubel V, Courageot MP, Desprès P. Dengue virus M protein contains a proapoptotic sequence referred to as ApoptoM. J Gen Virol. 2003;84:2781-93. http://dx.doi.org/10.1099/vir.0.19163-0
35. Desprès P, Flamand M, Ceccaldi PE, Deubel V. Human isolates of dengue type 1 virus induce apoptosis in mouse neuroblastoma cells. J Virol. 1996;70:4090-6.
36. Jan JT, Chen BH, Ma SH, Liu CI, Tsai HP, Wu HC, et al . Potential dengue virus-triggered apoptotic pathway in human neuroblastoma cells: Arachidonic acid, superoxide anion, and NF-kappaB are sequentially involved. J Virol. 2000;74:8680-91. http://dx.doi.org/10.1128/JVI.74.18.8680-8691.2000
37. Marianneau P, Flamand M, Deubel V, Despres P. Apoptotic cell death in response to dengue virus infection: The pathogenesis of dengue haemorrhagic fever revisited. Clin Diagn Virol. 1998;10:113-9.
38. Su HL, Lin YL, Yu HP, Tsao CH, Chen LK, Liu YT, et al . The effect of human bcl-2 and bcl-X genes on dengue virus-induced apoptosis in cultured cells. Virology. 2001;282:141-53. http://dx.doi.org/10.1006/viro.2000.0820
39. Lee CJ, Liao CL, Lin YL. Flavivirus activates phos-phatidylinositol 3-kinase signaling to block caspase-dependent apoptotic cell death at the early stage of virus infection. J Virol. 2005;79:8388-99. http://dx.doi.org/10.1128/JVI.79.13.8388-8399.2005
40. Long X, Li Y, Qi Y, Xu J, Wang Z, Zhang X, et al . XAF1 contributes to dengue virus-induced apoptosis in vascular endothelial cells. FASEB J. 2013;27:1062-73. http://dx.doi.org/10.1096/fj.12-213967
41. Chaturvedi UC. Tumour necrosis factor and dengue. Indian J Med Res. 2006;123:11-4.
42. Houghton N, Martín K, Giaya K, Rodríguez J, Bosch I, Castellanos JE. Comparación de los perfiles de transcripción de pacientes con fiebre de dengue y fiebre hemorrágica por dengue que muestra diferencias en la respuesta inmunitaria y claves en la inmunopatogénesis. Biomédica. 2010;30:587-97. http://dx.doi.org/10.7705/biomedica.v30i4.297
43. Sriram K, O´Callaghan JP. Divergent roles for tumor necrosis factor- a in the brain. J Neuroimmune Pharmacol. 2007;2: 140-53. http://dx.doi.org/1010.1007/s11481-007-9070-6
44. Deng M, Zhao JY, Ju XD, Tu PF, Jiang Y, Li ZB. Protective effect of tubuloside B on TNFalpha-induced apoptosis in neuronal cells. Acta Pharmacol Sin. 2004;25:1276-84.
45. Hopkins-Donaldson S, Bodmer JL, Bourloud KB, Brognara CB, Tschopp J, Gross N. Loss of caspase-8 expression in highly malignant human neuroblastoma cells correlates with resistance to tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis. Cancer Res. 2000;60:4315-9.
46. Eggert A, Grotzer MA, Zuzak TJ, Wiewrodt BR, Ho R, Ikegaki N, et al . Resistance to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in neuroblastoma cells correlates with a loss of caspase-8 expression. Cancer Res. 2001;61:1314-9.