COMUNICACIÓN BREVE
doi: http://dx.doi.org/10.7705/biomedica.v36i2.3116
1 Grupo de Investigaciones Biomédicas, Universidad de Sucre, Sincelejo, Colombia
2 Grupo BIOGEM, Universidad Nacional de Colombia, Medellín, Colombia
Contribución de los autores:
Luis Enrique Paternina: diseño del estudio, recolección de muestras y desarrollo de los análisis genéticos
Daniel Verbel-Vergara: recolección de muestras y ensayos moleculares
Eduar Elías Bejarano: diseño del estudio, coordinación del trabajo de laboratorio y análisis genéticos
Todos los autores participaron en la redacción del manuscrito.
Recibido: 12/11/15; aceptado: 22/02/16
Introducción. En las últimas décadas, el análisis de los genes mitocondriales se ha utilizado en los estudios poblacionales y filogenéticos de garrapatas, lo cual ha permitido numerosos avances en la sistemática de estos ácaros. El gen mitocondrial de la subunidad 16S del ARN ribosómico ( 16S ) es uno de los más usados, mientras que el gen mitocondrial de la citocromo oxidasa 1 ( COX1 ) se ha empleado recientemente y se propone como un marcador genético alternativo frente al 16S .
Objetivo. Evaluar la utilidad de los genes 16S y COX1 en los estudios genéticos de las garrapatas mediante el análisis de secuencias en tres especies de la región Caribe de Colombia.
Resultados. El análisis de secuencias mostró que los dos genes permitieron identificar las tres especies con mucha confiabilidad y con niveles de divergencia genética interespecífica relativamente similares (19 a 22 %), aunque solo el gen COX1 permitió detectar la variabilidad genética intraespecífica (hasta de ˜0,8 %). El análisis de saturación de sustituciones indicó que el gen 16S no se saturó con transiciones, mientras que el COX1 mostró saturación a partir de distancias de ˜17 %.
Conclusión. Los resultados indicaron que el gen 16S parece tener mejores características para los análisis filogenéticos interespecíficos dada su alta divergencia genética y baja saturación de transiciones, mientras que el gen COX1 parece ser más útil para estudios de variabilidad genética intraespecífica. Sin embargo, dado que el estudio se hizo a escala local, se requieren más investigaciones en diferentes escalas biogeográficas para establecer su utilidad en circunstancias más amplias y complejas.
Palabras clave: garrapatas, genética, ADN mitocondrial, filogenia, Colombia.
doi: http://dx.doi.org/10.7705/biomedica.v36i2.3116
Comparison of 16S and COX1 genes mitochondrial regions and their usefulness for genetic analysis of ticks (Acari: Ixodidae)
Introduction: In recent decades the analysis of mitochondrial genes has been used for population and phylogenetic studies of ticks allowing many advances in their systematics. Mitochondrial ribosomal 16S ( 16S ) subunit is one of the most frequently used among those genes available for tick analysis, whereas cytochrome oxidase gene 1 ( COX1 ) has recently been used and proposed as an alternative to the traditional 16S gene marker.
Objective: To evaluate the usefulness of 16S and COX1 in genetic studies of ticks by analyzing sequences of three species commonly found in the Caribbean region of Colombia.
Results: The analysis of both genes sequences allowed us to identify the three species with high levels of confidence and interspecific genetic divergence (19-22%), although only COX1 allowed us to detect intraspecific genetic variability (up to ˜0.8%). A substitution saturation analysis indicated that the 16S gene was not saturated with transitions while the COX1 gene showed saturation distances starting at ˜17%.
Conclusion: Our results indicated that the 16S gene seems to have better features for interspecific phylogenetic analyses because of its high level of genetic divergence and low saturation pattern, while the COX1 gene appears to be more useful for intraspecific genetic variability studies. However, as our study was conducted at a local scale, future studies at different biogeographical scales would help to establish its usefulness in wider and more complex scenarios.
Key words: ticks, genetics, DNA, mitochondrial, phylogeny, Colombia.
doi: http://dx.doi.org/10.7705/biomedica.v36i2.3116
Las garrapatas son ectoparásitos hematófagos obligados de un gran número de vertebrados que tienen gran importancia veterinaria y médica por su papel en la transmisión de numerosas enfermedades (1). Entre las garrapatas más conocidas se encuentran las de la especie Rhipicephalus microplus , las cuales ocasionan enormes pérdidas económicas por los daños directos e indirectos que ocasiona en el ganado bovino (2). También están las garrapatas del complejo R. sanguineus, vectores de la erliquiosis y la trombocitopenia cíclica infecciosa en caninos (3,4), que también pueden transmitir enfermedades a los humanos, como la fiebre botonosa mediterránea (2) y la fiebre manchada de las Montañas Rocosas (5).
Clásicamente, la taxonomía y la sistemática de las garrapatas se han definido mediante estudios morfológicos; sin embargo, en las últimas décadas las herramientas moleculares han tenido un gran impacto en la sistemática de este importante grupo de artrópodos. Entre los ejemplos más notables de ajustes taxonómicos en los que los análisis genéticos han sido definitivos en la descripción de nuevas especies y en la identificación de complejos en las familias Ixodidae y Argasidae, están el complejo Rhipicephalus sanguineus (6-8), el grupo Ornithodoros talaje (9) y el complejo Amblyomma cajennense (10,11). Debido a las peculiaridades biológicas y ecológicas que presentan las diferentes especies y grupos de especies de garrapatas, los estudios genéticos se han convertido en una herramienta importante para explorar las relaciones poblaciones (8,12-14), delimitar las especies (15-17), hacer los análisis filogenéticos (18-20), e, incluso, identificar potenciales zonas de riesgo de transmisión de enfermedades (3,7,8).
Ante este auge de las metodologías moleculares para el estudio de diversos aspectos de la ecología y la taxonomía de las garrapatas (7), es necesario conocer los beneficios y limitaciones de los marcadores moleculares utilizados para tales fines. Entre dichos marcadores se encuentran las secuencias mitocondriales de la subunidad 16S del ARN ribosómico (16S), la subunidad 12S del ARN ribosomal (12S), la región D-loop, el gen de la citocromo oxidasa I ( COX1 ), el gen de la citocromo oxidasa II ( COX2 ), la subunidad 5 de la deshidrogenasa de NADH (NAD5) y regiones nucleares como el espaciador interno transcrito 2 (ITS2), el gen 18S ribosómico, el gen de la subunidad grande ribosómica ( LSU ), el gen de la ARN polimerasa II ( RNApol II ) y el gen de la gliceraldehído-3-fosfato deshidrogenasa ( G3PDH ) (10,14-18,20,21-26).
Entre las secuencias mencionadas, el gen 16S es uno de los más utilizados en los estudios de taxonomía y variabilidad genética de este grupo de artrópodos y, recientemente, algunos autores han empleado el gen mitocondrial COX1 de la iniciativa de código de barras de ADN ( Barcode of Life , www.barcodeoflife.org) (27), con la cual se aspira a identificar y reconocer la diversidad de especies a nivel global con base en la variabilidad genética que exhiben regiones del gen COX1 en diversos grupos de artrópodos (16,26,28).
Dadas las múltiples opciones que existen para el trabajo en taxonomía o filogenética de las garrapatas y el creciente uso de las herramientas moleculares en la investigación sobre las enfermedades transmitidas por estas en Colombia, el objetivo del presente estudio fue evaluar la utilidad de los genes mitocondriales 16S y COX1 en los estudios genéticos de las garrapatas duras a escalas geográficas locales.
Materiales y métodos
Áreas de estudio y garrapatas
Las garrapatas se recolectaron mediante la búsqueda en animales domésticos provenientes de cinco municipalidades pertenecientes a cuatro subregiones ecológicas del departamento de Sucre: Sincelejo (SinSP, subregión Montes de María), Sampués (SamCM, subregión Sabanas), Corozal (CorL, subregión Sabanas), Toluviejo (TolC, subregión golfo del Morrosquillo) y San Marcos (SanM, subregión San Jorge).
A pesar de su cercanía geográfica, con distancias lineales entre 9 y 92 km, estas localidades presentan diferencias considerables en cuanto al tipo y la densidad de la vegetación, la topografía, la hidrografía y las actividades económicas que eventualmente podrían influir en la variabilidad genética de las garrapatas (la información adicional sobre las subregiones puede consultarse en www.sucre.gov.co). Para el estudio se seleccionaron individuos sin alimentar del complejo R. sanguineus , R. microplus y Dermacentor nitens Extracción de ácidos nucleicos y amplificación del ADN
Las garrapatas se trituraron en nitrógeno líquido y la extracción de los ácidos nucleicos se hizo utilizando el estuche Qiamp DNA Mini Kit ™ (Qiagen, Hilden, Germany), según las instrucciones del fabricante. Los extractos de ADN de cada muestra se amplificaron utilizando los genes mitocondriales 16S , el cual genera un fragmento de ˜360 pb, y COX1 , que genera un fragmento de ˜710 pb, (18,28). La amplificación de los dos genes se hizo con la siguiente mezcla de reacción: 1,5 mM de MgCl 2 , 3 µl del extracto de ADN, 15 pmol de cada cebador y 1 U de Taq Platinum de Invitrogen (Life Technologies). Los amplicones resultantes se secuenciaron mediante electroforesis capilar en un secuenciador ABI 3730XLs (Applied Biosystems).
Comparación de marcadores genéticos
Solo los individuos en los que fue posible la amplificación de ambos genes mitocondriales, 16S (˜360 pb) y COX1 (˜710 pb), se sometieron a la secuenciación directa del ADN; según la disponibilidad de estas muestras, se incluyó al menos un individuo de cada localidad para cada taxón analizado.
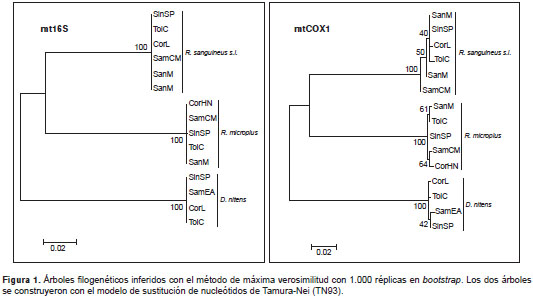
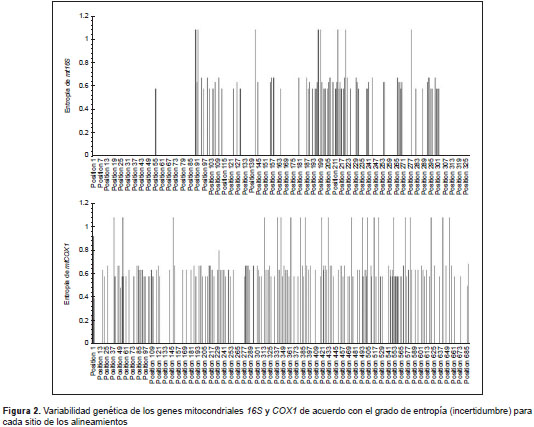
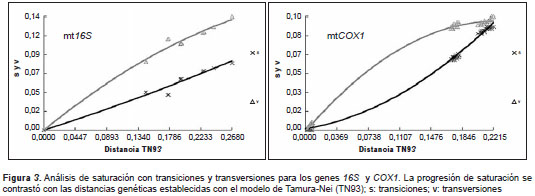
Los electroforegramas obtenidos para los genes 16S y COX1 se editaron manualmente con el programa Chromas Lite; se obtuvieron las secuencias de consenso por muestra y se alinearon utilizando el algoritmo de Clustal W, con lo cual se generó un alineamiento múltiple de 328 sitios para las secuencias del gen 16S y de 690 sitios para las del gen COX1 . Una vez obtenido el alineamiento, se procedió a estimar el mejor modelo de sustitución de nucleótidos mediante el JModelTest y con el criterio de información Akaike corregido (AICc) (29). Los árboles filogenéticos se analizaron con el algoritmo de máxima verosimilitud de 1.000 repeticiones con el método de bootstrap y el mejor modelo de sustitución de nucleótidos calculado previamente con el programa MEGA 5.2 (30). Además, se valoró el grado de saturación de las sustituciones de los marcadores genéticos mediante un gráfico de saturación con transiciones (s) y transversiones (v), así como la distribución de los sitios variables y el grado de variabilidad genética por sitio a lo largo de las matrices de secuencias de ambos genes mediante un análisis de entropía en el programa DAMBE 5 (31).
También, se calcularon las distancias intraespecíficas e interespecíficas para establecer una comparación entre los genes en el análisis hecho con la herramienta MEGA 5.2, y se calcularon los estimadores de diversidad haplotípica (Hd), el número medio de diferencias pareadas entre nucleótidos (k) y la diversidad de los nucleótidos (p) obtenidos con el programa DnaSP, versión 5 (32).
Resultados
Se seleccionaron 15 de las 50 garrapatas analizadas para la secuenciación con ambos genes: seis del complejo R. sanguineus , cinco de R. microplus y cuatro de D. nitens . Los haplotipos del gen 16S se encuentran depositados en GenBank bajo los códigos KT906174-KT906176 y los haplotipos encontrados del gen COX1 bajo los códigos KT906177-KT906190.
En los árboles filogenéticos se observó que, como era de esperarse, ambos marcadores genéticos eran capaces de diferenciar con gran confiabilidad las tres especies de garrapatas (figura 1), aunque las distancias interespecíficas medias según el modelo de Tamura-Nei con el COX1 siempre fueron mayores a las del gen mitocondrial 16S (cuadro 1).
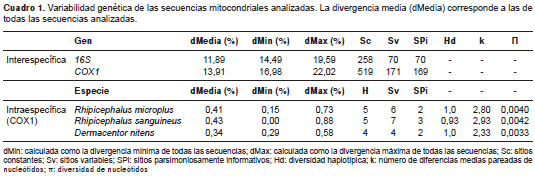
Las distancias promedio totales fueron mayores cuando se utilizó el marcador COX1 (13,91 %) en comparación con el marcador 16S (11,89 %). Por otro lado, el rango de distancia interespecífica, calculado como la diferencia porcentual entre la distancia máxima (dMax) y la distancia mínima (dMin) de todas las secuencias de cada marcador mitocondrial, indicó que el gen 16S proveía un mayor rango de distancias, con 5,1 %, mientras que el COX1 fue similar aunque inferior, con 5,04 %. Con respecto al análisis de saturación (figura 3), a diferencia del gen COX1 , el cual presentó saturación con transversiones a partir de distancias Tamura-Nei de ˜17 %, el marcador 16S no se saturó con sustituciones. Sin embargo, la remoción de la tercera posición del codón en el análisis de saturación del gen COX1 permitió superar este inconveniente, aunque la variabilidad máxima observada con las dos primeras posiciones del codón fue de hasta 6,24 % (no se presentan los datos).
En cuanto a la variabilidad intraespecífica, el gen 16S produjo un solo haplotipo de cada especie analizada, y se estimó una distancia genética de cero a nivel intraespecífico, lo cual contrasta claramente con los 14 haplotipos detectados en las 15 muestras analizadas con el gen COX1 , el cual presentó distancias intraspecíficas de entre 0,34 y 0,43 % (cuadro 1). La variabilidad intraespecífica (dMax-dMin) detectada con el marcador genético COX1 , se relacionó con el número de muestras o con la especie de garrapata analizada; en cuatro individuos de D. nitens la variabilidad fue de 0,29%; en cinco individuos de R. microplus la variabilidad fue de 0,58 % y para seis individuos de R. sanguineus la variabilidad genética fue de 0,88 %. En cuanto a los parámetros de diversidad genética k y p, quedó claro que el grado de diversidad genética detectable también guardó una estrecha relación con el tamaño de la muestra.
Discusión
Dada su gran importancia económica, los estudios sobre garrapatas en Colombia se han enfocado básicamente en su impacto en el sector productivo y en la identificación de las especies involucradas en los brotes de enfermedades transmitidas por ellas (33,34). En los últimos años se ha reconocido la existencia de múltiples complejos de especies en Latinoamérica y el papel que pueden tener algunos de ellos en la dinámica epidemiológica de enfermedades de interés médico y veterinario en la región (3,4,7,8,10,11). Ante este panorama, los análisis genéticos de las garrapatas pueden proveer información relevante para el estudio y la vigilancia de las enfermedades transmitidas por garrapatas en nuestro país.
En la presente evaluación de los marcadores genéticos mitocondriales 16S y COX1 , el gen 16S fue especialmente útil para la separación de especies debido a su baja variabilidad genética intraespecífica, lo cual facilita la asignación de especies y da un rango de distancia interespecífica ligeramente mayor, por lo que se sugiere que este marcador sea utilizado en los estudios sobre relaciones interespecíficas. Por otro lado, aunque el marcador COX1 también permite diferenciar con gran confiabilidad las tres especies de garrapatas analizadas, fue evidente que es más útil para detectar una variabilidad genética intraespecífica de hasta 0,88 % en muestras que no presentaron variabilidad genética con el gen 16S .
Por otro lado, el análisis de retención de la información filogenética mostró que el marcador 16S no se saturaba con sustituciones a la escala taxonómica analizada en el presente trabajo, lo que respalda los hallazgos de otros autores que resaltan la utilidad de esta secuencia mitocondrial para filogenias en la escala analizada y para estudios de taxonomía molecular (18,21,35,36). Por el contrario, el análisis con al gen COX1 mostró que este marcador se saturó a partir de distancias calculadas con el método de Tamura-Nei de ˜17 %, lo que limita su utilidad filogenética aparente a los estudios intraespecíficos o a los análisis genéticos de complejos de especies relativamente incipientes, tal como lo han señalado otros autores en otros grupos de ácaros (36,37). Para reducir la saturación del gen COX1 con el fin de utilizarlo en análisis filogenéticos interespecíficos, se recomienda la remoción de la tercera posición del codón (como se hizo en este caso). Sin embargo, esto reduce notoriamente la variabilidad genética del fragmento analizado y, a pesar de estos ajustes, es posible que sigan presentándose homoplasias que reduzcan la confiabilidad de los resultados, tal como ha sucedido en trabajos previos en ácaros de la familia Tetranychidae (37).
En conclusión, los dos marcadores genéticos evaluados presentan características diferentes que permitieron obtener una visión complementaria de las relaciones genéticas entre taxones de garrapatas y dentro de ellos. Por esta razón, se recomienda tener en cuenta las características propias de cada gen en futuros estudios genéticos.
Por último, dado que el presente trabajo se hizo con muestras obtenidas a partir de poblaciones cercanas de las tres especies de garrapatas, los estudios futuros en los que se comparen estos dos marcadores genéticos a escalas geográficas mayores permitirán establecer la variabilidad y la utilidad de ambas secuencias en escenarios más amplios y complejos.
Al programa de Jóvenes Investigadores e Innovadores del Departamento Administrativo de Ciencia, Tecnología e Innovación de Colombia (Colciencias), por el apoyo a Daniel Verbel-Vergara, y al programa de Becas Doctorales Nacionales de la misma entidad (Convocatoria 528/2011), por el apoyo a Luis Enrique Paternina. Agradecemos igualmente a Francisco Javier Díaz del Grupo de Inmunovirología de la Universidad de Antioquia, por sus recomendaciones en el análisis de la saturación de sustituciones, así como a los evaluadores anónimos del manuscrito, cuyos comentarios y críticas constructivas permitieron mejorarlo significativamente.
No existe conflicto de intereses para declarar.
Colciencias, proyecto 112951929253 (Convocatoria 519/2010), y Dirección de Investigación de la Universidad Nacional de Colombia, sede Medellín, proyecto 20810 (código QUIPU 201010013308).
Correspondencia: Luis E. Paternina, Carrera 14 N° 16B-32, barrio La Pajuela, Sincelejo, Colombia Teléfono: (575) 282 0830 luispaterninat@gmail.com
1. Baneth G. Tick-borne infections of animals and humans: A common ground. Int J Parasitol. 2014;44:591-6. http://dx.doi.org/10.1016/j.ijpara.2014.03.011
2. Jongejan F, Uilenberg G. The global importance of ticks. Parasitology. 2004;123:S3-14.
3. Moraes-Filho J, Krawczak F, Costa FB, Soares JF, Labruna MB. Comparative evaluation of the vector competence of four South American populations of the Rhipicephalus sanguineus group for the bacterium Ehrlichia canis , the agent of canine monocytic ehrlichiosis. PLoS One. 2015;10:e0139386. http://dx.doi.org/10.1371/journal.pone.0139386
4. Cicuttin G, Tarragona EL, De Salvo MN, Mangold A, Nava S. Infection with Ehrlichia canis and Anaplasma platys (Rickettsiales: Anaplasmataceae) in two lineages of Rhipicephalus sanguineus sensu lato (Acari: Ixodidae) from Argentina. Ticks Tick Borne Dis. 2015;6:724-9. http://dx.doi.org/10.1016/j.ttbdis.2015.06.006
5. Demma L, Traeger M, Nicholson W, Paddock C, Blau D, Eremeeva M, et al . Rocky Mountain spotted fever from an unexpected tick vector in Arizona. N Eng J Med. 2005;353:587-94. http://dx.doi.org/10.1056/NEJMoa050043
6. Szabó MPJ, Mangold AJ, João CF, Bechara GH, Guglielmone AA. Biological and DNA evidence of two dissimilar populations of the Rhipicephalus sanguineus tick group (Acari: Ixodidae) in South America. Vet Parasitol. 2005;130:131-40.
7. Moraes-Filho J, Marcili A, Nieri-Bastos FA, Richtzenhain LJ, Labruna MB. Genetic analysis of ticks belonging to the Rhipicephalus sanguineus group in Latin America. Acta Trop. 2011;117:51-5. http://dx.doi.org/10.1016/j.actatropica.2010.09.006
8. Nava S, Mastropaolo M, Venzal JM, Mangold AJ, Guglielmone AA. Mitochondrial DNA analysis of Rhipicephalus sanguineus sensu lato (Acari: Ixodidae) in the Southern Cone of South America. Vet Parasitol. 2012;190:547-55. http://dx.doi.org/10.1016/j.vetpar.2012.06.032
9. Venzal JM, Estrada-Peña A, Mangold AJ, González-Acuña D, Guglielmone AA. The Ornithodoros (Alectorobius) talaje species group (Acari: Ixodida: Argasidae): Description of Ornithodoros ( Alectorobius ) rioplatensis n. sp. from southern South America. J Med Entomol. 2008;45:832-40. http://dx.doi.org/10.1093/jmedent/45.5.832
10. Beati L, Nava S, Burkman EJ, Barros-Battesti DM, Labruna MB, Guglielmone AA, et al . Amblyomma cajennense (Fabricius, 1787) (Acari: Ixodidae), the Cayenne tick: Phylogeography and evidence for allopatric speciation. BMC Evol Biol. 2013;13:267. http://dx.doi.org/10.1186/1471-2148-13-267
11. Nava S, Beati L, Labruna MB, Cáceres AG, Mangold AJ, Guglielmone AA. Reassessment of the taxonomic status of Amblyomma cajennense with the description of three new species, Amblyomma tonelliae n. sp., Amblyomma interandinum n. sp. and Amblyomma patinoi n. sp., and reinstatement of Amblyomma mixtum , and Amblyomma sculptum . Ticks Tick Borne Dis. 2014;5:252-76. http://dx.doi.org/10.1016/j.ttbdis.2013.11.004
12. Trout RT, Steelman CD, Szalanski AL. Population genetics of Amblyomma americanum (Acari: Ixodidae) collected from Arkansas. J Med Entomol. 2010;47:152-61. http://dx.doi.org/10.1093/jmedent/47.2.152
13. Beati L, Patel J, Lucas-Williams H, Adakal H, Kanduma EG, Tembo-Mwase E, et al . Phylogeography and demographic history of Amblyomma variegatum (Fabricius) (Acari: Ixodidae), the tropical bont tick. Vector Borne Zoonotic Dis. 2012;12:514-25. http://dx.doi.org/10.1089/vbz.2011.0859
14. Radulovi c Ž, Milutinovi c M, Tomanovi c S, Mulenga A. Exon variability of gene encoding glycerol-3-phosphate dehydrogenase of Ixodes ricinus ticks. Parasite. 2010; 17:363-8.
15. Marrelli MT, Souza LF, Marques RC, Labruna MB, Matioli SR, Tonon AP, et al . Taxonomic and phylogenetic relationships between neotropical species of ticks from genus Amblyomma (Acari: Ixodidae) inferred from second internal transcribed spacer sequences of rDNA. J Med Entomol. 2007;44:222-8. http://dx.doi.org/10.1093/jmedent/44.2.222
16. Latrofa MS, Dantas-Torres F, Annoscia G, Cantacessi C, Otranto D. Comparative analyses of mitochondrial and nuclear genetic markers for the molecular identification of Rhipicephalus spp. Infect Genet Evol. 2013;20:422-7. http://dx.doi.org/10.1016/j.meegid.2013.09.027
17. Lv J, Wu S, Zhang Y, Chen Y, Feng C, Yuan X, et al . Assessment of four DNA fragments (COI, 16S rDNA, ITS2, 12S rDNA) for species identification of the Ixodida (Acari : Ixodida). Parasit Vectors. 2014;7:93. http://dx.doi.org/10.1186/1756-3305-7-93
18. Black WC, Piesman J. Phylogeny of hard- and soft-tick taxa (Acari: Ixodida) based on mitochondrial 16S rDNA sequences. Proc Natl Acad Sci USA. 1994;91:10034-8.
19. Crampton A, McKay I, Barker SC. Phylogeny of ticks (Ixodida) inferred from nuclear ribosomal DNA. Int J Parasitol. 1996;26:511-7.
20. Mangold AJ, Bargues MD, Mas-Coma S. Mitochondrial 16S rDNA sequences and phylogenetic relationships of species of Rhipicephalus and other tick genera among Metastriata (Acari: Ixodidae). Parasitol Res. 1998;84:478-84.
21. Nava S, Guglielmone AA, Mangold AJ. An overview of systematics and evolution of ticks. Front Biosci. 2009;14:2857-77. http://dx.doi.org/10.2741/3418
22. Leo SST, Pybus MJ, Sperling FA. Deep mitochondrial DNA lineage divergences within Alberta populations of Dermacentor albipictus (Acari: Ixodidae) do not indicate distinct species. J Med Entomol. 2010;47:565-74. http://dx.doi.org/10.1093/jmedent/47.4.565
23. Anstead CA, Krakowetz CN, Mann AS, Sim KA, Chilton NB. An assessment of genetic differences among ixodid ticks in a locus within the nuclear large subunit ribosomal RNA gene. Mol Cell Probes. 2011;25:243-8. http://dx.doi.org/10.1016/j.mcp.2011.06.002
24. Beati L, Keirans JE. Analysis of the systematic relationships among ticks of the genera Rhipicephalus and Boophilus (Acari: Ixodidae) based on mitochondrial 12S ribosomal DNA gene sequences and morphological characters. J Parasitol. 2001;87:32-48. http://dx.doi.org/10.1645/0022-3395(2001)087[0032:AOTSRA]2.0.CO;2
25. Fang Q, Keirans J, Mixson T. The use of the nuclear protein-encoding gene, RNA polymerase II, for tick molecular systematics. Exp Appl Acarol. 2002;28:69-75.
26. Chitimia L, Lin R-Q, Cosoroaba I, Wu X-Y, Song H-Q, Yuan Z-G, et al . Genetic characterization of ticks from southwestern Romania by sequences of mitochondrial COX1 and nad5 genes. Exp Appl Acarol. 2010;52:305-11. http://dx.doi.org/10.1007/s10493-010-9365-9
27. Ratnasingham S, Hebert PD. Bold: The Barcode of Life Data System (www.barcodinglife.org). Mol Ecol Notes. 2007;7:355-64. http://dx.doi.org/10.1111/j.1471-8286.2007.01678.x
28. Hebert PD, Cywinska A, Ball SL, deWaard JR. Biological identification through DNA barcodes. Proc R Soc Lond B. 2003;270:313-21. http://dx.doi.org/10.1098/rspb.2002.2218
29. Posada D. jModelTest: Phylogenetic model averaging. Mol Biol Evol. 2008;25:1253-6. http://dx.doi.org/10.1093/molbev/msn083
30. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28: 2731-9. http://dx.doi.org/10.1093/molbev/msr121
31. Xia X. DAMBE5: A comprehensive software package for data analysis in molecular biology and evolution. Mol Biol Evol. 2013;30:1720-8. http://dx.doi.org/10.1093/molbev/mst064
32. Librado P, Rozas J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009; 25:1451-2. http://dx.doi.org/10.1093/bioinformatics/btp187
33. Hidalgo M, Orejuela L, Fuya P, Carrillo P, Hernández J, Parra E, et al . Rocky mountain spotted fever, Colombia. Emerg Infect Dis. 2007;13:1058-60. http://dx.doi.org/10.3201/eid1307.060537
34. Cortés-Vecino JA, Betancourt JA, Argüelles J, Pulido LA. Distribución de garrapatas Rhipicephalus microplus en bovinos y fincas del altiplano cundiboyacense. Corpoica Cienc Tecnol Agropecu. 2010;11:73-84.
35. Crosbie PR, Boyce WM, Rodwell TC. DNA sequence variation in Dermacentor hunteri and estimated phylogenies of Dermacentor spp. (Acari: Ixodidae) in the New World. J Med Entomol. 1998;35:277-88. http://dx.doi.org/10.1093/jmedent/35.3.277
36. Cruickshank RH. Molecular markers for the phylogenetics of mites and ticks. Sys Applied Acarol. 2002;7:3-14.
37. Navajas M, Gutiérrez J, Lagnel J, Boursot P. Mitochondrial cytochrome oxidase I in Tetranychid mites: A comparison between molecular phylogeny and changes of morphological and life history traits. Bull Entomol Res. 1996;86:407-17.