Biomédica 2017;37(Supl.2):143-54
doi: https://doi.org/10.7705/biomedica.v34i2.3452
ORIGINAL ARTICLE
1 Grupo de Investigación en Sistemática Molecular, Escuela de Biociencias, Facultad de Ciencias, Universidad Nacional de Colombia, Medellín, Colombia
2 Grupo de Genética Molecular, GENMOL, Universidad de Antioquia, Medellín, Colombia
3 Programa de Estudio y Control de Enfermedades, PECET, Universidad de Antioquia, Medellín, Colombia
Author’s contributions:
Andrés López-Rubio: DNA sequences, phylogenetic analyses, secondary structure modeling and drafting of the manuscript
Juan David Suaza: Field work, specimens breeding and taxonomic determination
Charles Porter: Counseling on selection of collection sites, verification of specimens taxonomic determinations
Sandra Uribe: Counseling on molecular biology experiments, DNA sequence analyses and manuscript organization
Gabriel Bedoya: Counseling on secondary structure modelling of the SertRNA mitochondrial gene
Iván Darío Vélez: Counseling on the selection of field collection sites and the establishment of controlled breeding conditions
All authors contributed to the critical review of the manuscript.
Received: 12/08/16; accepted: 15/08/17
Introduction: Mitochondrial DNA has proven its utility for the study of insect evolution. Genes such as cytochrome b (Cytb) and the transfer gene for serine (SertRNA) can be used to compare closely related organisms.
Objective: The phylogenetic utility of Cytb-SertRNA-IG1-ND1 was tested for polymorphisms, and secondary structure modeling in SertRNA was done to detect possible cryptic species in Anopheles neivai.
Materials and methods: Specimens from Colombia, Guatemala, and the type locality in Panamá were collected and sequenced for specimen comparison based on DNA polymorphisms, and secondary structure modeling for the SertRNA gene.
Results: Thirty-six sequences for A. neivai and A. pholidotus were obtained.
Conclusions: Polymorphic variants were detected in A. neivai for Cytb-SertRNA-IG1- ND1. Despite this variation in A. neivai, cryptic species could not be detected.
Key words: Anopheles; malaria; RNA, transfer; DNA, mitochondrial; Colombia; Panamá; Guatemala.
doi: https://doi.org/10.7705/biomedica.v34i2.3452
Señal filogenética de la región Cytb-SertRNA -IG1-ND1 en Anopheles (Kerteszia) neivai Howard, Dyar & Knab, 1913
Introducción. El ADN mitocondrial ha demostrado su utilidad para el estudio de la evolución en los insectos. Existen algunos genes mitocondriales como el citocromo b (Cytb) y el gen de transferencia para el aminoácido serina (SertRNA) que pueden usarse en el diagnóstico de especies estrechamente relacionadas.
Objetivo. Explorar la utilidad filogenética de la región Cytb-SertRNA-IG1-ND1 para detectar posibles especies crípticas en Anopheles neivai.
Materiales y métodos. Se recolectaron especímenes en Colombia, Guatemala y en la localidad tipo en Panamá, los cuales se secuenciaron y se compararon mediante el polimorfismo de ADN en toda la región y mediante la simulación de estructuras secundarias del gen SertRNA.
Resultados. Se obtuvieron las secuencias de especímenes de A. neivai (34) y A. pholidotus (2).
Conclusiones. Se detectaron algunos polimorfismos para la regiónCytb-SertRNA-IG1-ND1 en A. neivai, pero no así especies crípticas.
Palabras clave: Anopheles; malaria; ARN de transferencia; ADN mitocondrial; Colombia; Panamá; Guatemala.
doi: https://doi.org/10.7705/biomedica.v34i2.3452
Anopheles neivai Howard, Dyar & Knab, 1913, is distributed from the Yucatán Peninsula (México) to the Guayas province (Ecuador) (1). In Colombia, it has been recognized as a secondary malaria vector in the Pacific Coast with local importance in small towns such as Charambirá, Santa Bárbara Iscuandé and Buenaventura (2-4). There are also reports of naturally infected females with Plasmodium falciparum in Buenaventura (5). As regards its biology, immature stages develop in bromeliads at mangrove (1). Activity in adults occurs at dawn and dusk (2) when younger females prefer to feed (6) and can be found in homes and in their surroundings (3,7). There are also biting reports in mangrove environments and in boats during fishing activities, which increase the epidemiological risk for local habitants (2).
Anophelines are recognized as a complex group, where it is common to find complexes of cryptic species (8). In Neotropical Anopheles, there are recognized cryptic species complexes that have importance as malaria vectors such as A. triannulatus (9) and A. albitarsis (10). For A. neivai, previous research suggests the existence of a species complex (2,11) based on morphological polymorphism and exploratory information on molecular variability (12).
The use of mitochondrial DNA (mtDNA) has played a vital role in understanding the phylogenetic relationships in Anopheles, including diversification patterns, divergence time estimation (13,14) and the recognition of cryptic species complexes (15-17). There are several advantages to using mtDNA, including variable mutation rates in eukaryotes, lack of introns, and high abundance of copies, which makes mtDNA relatively easy to obtain even from degraded samples (18). In contrast, phylogenetic signals of mtDNA can be altered by mitochondrial introgression (19) and by indirect selection and linkage disequilibrium caused by maternally inherited symbionts in arthropods (20). For phylogeny studies, fragments of cytochrome oxidase I gene (COI), have been extensively used (21-23). However, other genes in the mtDNA genome can be useful for reconstructing evolutionary relationships, including those involved in the respiratory chain complex I and III such as NADH dehydrogenase subunit I (ND1) and cytochrome b (Cytb) (24,25).
In addition, the transfer RNA genes (tRNA), also present in the mitochondrial genome, have a pivotal role in protein biosynthesis (26). The critical function of these genes limits the mutations they can accommodate, which is a desirable tool for studying evolution among closely related organisms. This is particularly important where selective pressure, mutation bias and genetic drift become relevant factors in modeling the codon use for each species (27,28). Furthermore, comparative analysis of the secondary structure of these genes improves the quality of alignments and, thereby, leads to more robust phylogenetic inferences (29,30).
In this work, we explored the phylogenetic utility of a mitochondrial region with fragments of Cytb, ND1 and the secondary structure models of serine transfer RNA (SertRNA) variants to differentiate specimens identified morphologically as A. neivai.
Materials and methods
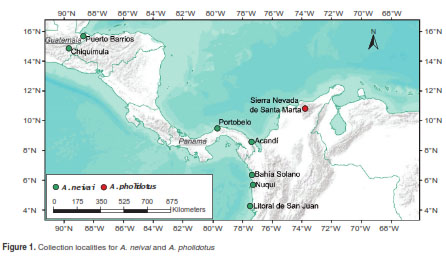
Specimens from Kerteszia were collected from six localities in Chocó, Colombia: Bahía Solano (Playa Potes and Nabugá), Litoral de San Juan (Charambirá), Acandí, Nuquí and Jurubidá, and the Caribbean region near the Sierra Nevada de Santa Marta. In Central America, the type locality for A. neivai at Portobelo (Panamá) was sampled, as well as two localities in Guatemala: Puerto Barrios and Chiquimula (figure 1, table 1). All the immature stages were collected in bromeliads using manual pipetting and reared under laboratory conditions to obtain adults (31), while field adults were collected using Shannon traps. Specimen identification was performed with dichotomous identification keys (32,33).
For DNA extraction, two legs were removed from each adult, whereas partial abdomens were used for the larval stages. The DNA extraction procedure for all specimens was based on a grind buffer protocol (34). The subsequent PCR conditions for the Cytb-tRNASer-IG1-ND1 region were as follows: 17.1 µL (dH2O); 6 µL (buffer 5X); 2 µL (dntp [2.5 mM]); 1 µL (MgCl2 [25 mM]); 0.3 µL Gotaq® Flexi (Promega); and 1 µL [10 mM] for primers CB3CF (CAYATTCAACCWGAATGATA) and NINFRGGT AYWTTGCCTCGAWTTCGWTATGA) (35). Primer sequences were modified from previously used primers CB-J-11338 and NI-N-11841 (36). The PCR reactions used the following conditions: 94°C/30 s, 47°C/30 s, 72°C/60 s. All the PCR experiments were performed on a PTC-100 BioRad ® thermal cycler. Subsequent fragments were examined by agarose gel electrophoresis (1%). Successive positive PCR fragments were sequenced using an ABI 3500XL ® (Applied Biosystems) automated capillary electrophoresis sequencer at the Centers for Disease Control and Prevention (CDC), Atlanta.
Consensus sequences were assembled using Geneious 6.0 (37). Sequences were checked against available records in the National Center for Biotechnology Information (NCBI) (http://www.ncbi.nlm.nih.gov/) using the BLAST program (38). In addition, the sequences were verified for possible mitochondrial copies at the nucleus (NUMT), following the procedure suggested by Hlaing, et al. (39). The sequences of each region were aligned using Clustal X 2 (40) and included reference sequences of A. albitarsis (NC_020662) from NCBI.
The utility of this region was first assessed by identifying polymorphic sites supporting variants between specimens of A. neivai with other specimens from the subgenus Kerteszia and A. albitarsis (subgenus Nyssorhynchus) using DnaSP 5 (41). Then, tRNASer secondary structures for each variant were modeled using the RNA Fold package available in Vienna RNA 2.0 (42). Parameters were based on Mathews, et al. (43) using minimum free energy and partition, and no GU pairs at the end of helices, allowing dangling energies on both sides of the helix at 37 °C. The phylogenetic signal of this region was estimated using maximum parsimony (MP) with bootstrap resampling using 100,000 replicates through MEGA 7 (44), and a maximum likelihood (ML) with bootstrap support of 1,000 replicates. A Generalized Time Reversible model GTR (45) was implemented using PhyML (46), and Bayesian inference (BI) under GTR model, assuming relaxed clock rates and Yule speciation process for BI (47) using BEAST 2.4 (48).
Results
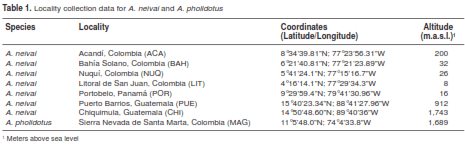
Adults from A. neivai, including topotypic specimens from Portobelo (Panamá), were identified based on the following set of characters from (33): mesanepimeron with an upper row seta (figure 2A), abdominal terguites I to IV without dark decumbent scales (figure 2B), acrostical area without white setae (figure 2C), and scale patterns on hind tarsomers (figure 2D). Additionally, specimens from immature stages were identified using the VIth abdominal segment and C setae from VIIth abdominal segment described in (32). No variability for the two immature specimens of A. pholidotus was observed.
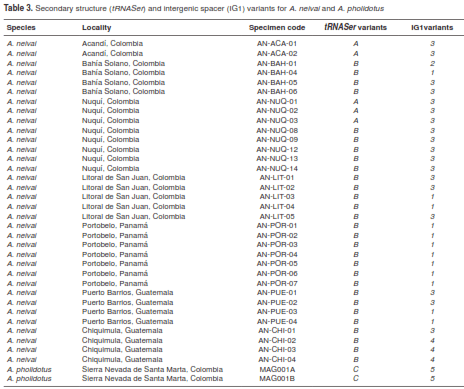
The entire set of specimens produced a total of 36 sequences (table 2): 34 for A. neivai and two for A. pholidotus. Consequently, the amplified fragment of 411 bp consisted of 304 bp for Cytb, 68 bp for tRNASer, 25 bp for IG1, and 11 bp for ND1. Among them, 51 polymorphisms were identified for Cytb, six for tRNASer (including 2 indels), eight for IG1 (including 5 indels) and four for ND1.
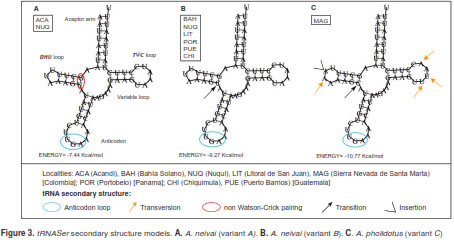
Three variant secondary structures were associated with tRNASer polymorphisms (table 3). The first two structures (A and B) were found with A. neivai specimens, and the other (C) was found in A. pholidotus. In the A. neivai variants, the only difference is located at the dihydrouridine loop (DHU) as a result of a base substitution (U → C) (figure 3). When comparing A. neivai and A. pholidotus further differences were evident, including a length difference in the DHU loop as a consequence of an adenine insertion, followed by a transversion in A. pholidotus. All the remaining differences between A. neivai and A. pholidotus consisted of transversions in the pseudouridine loop (TøC) as synonymous substitutions detected in A. pholidotus (A → U, A → U and U → A).
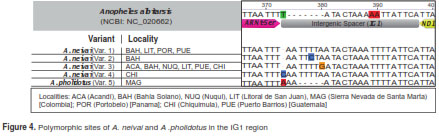
The intergenic spacer (IG1) between tRNASer and ND1 exhibited differences in length. A larger intergenic spacer was present in most of the specimens of A. neivai (24-25 bp), in contrast to A. pholidotus (20 bp) and A. albitarsis (18). The nucleotide variability in this region for A. neivai consisted of four variants, base substitutions found in variants 1 to 3, and an insertion on position 373 for variant 4 (Chiquimula, Guatemala). The same insertion was found in A. pholidotus (figure 4).
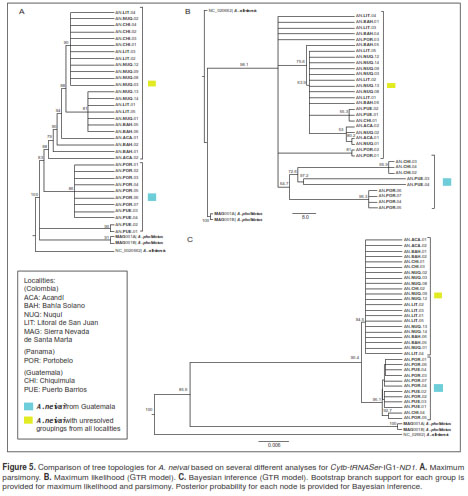
The comparison of three different approaches (MP, ML and IB) revealed similar patterns. One group with unresolved specimens for the majority of localities and another group including specimens from Panamá and Guatemala (figure 5). The overall pattern of nucleotide substitution estimated by ML across the Cytb-tRNASer-IG1-ND1 regions recovered from several trees revealed phylogenetic signal for the Cytb fragment and tRNASer gene (figure 5b). Equivalent groupings based on trees that included both fragments (figure 6a and 6b), were evident in specimens of A. pholidotus from Colombia and in A. neivai from Central America (Panamá and Guatemala localities). In contrast, when these regions were excluded, leaving only the IG1ND1 fragment (figure 6c), the relationships between A. neivai specimens in the Colombian localities and those from Central America were impossible to resolve due to the presence of polytomies.
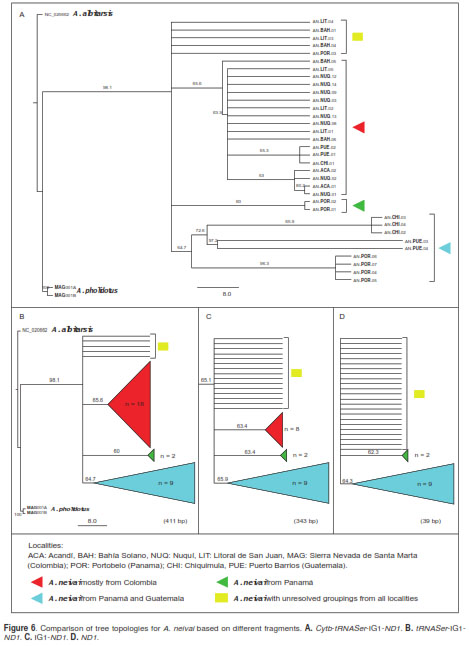
Discussion
Phylogenetic signal is the tendency of closely related species to resemble one another (49). It is also a property of stochastic evolution along a hierarchical tree, depending on sample size, mutational rate and branch length (50). Therefore, testing the phylogenetic signal of a molecular marker is essential for phylogeny reconstruction of closely related and cryptic species, before proposing evolutionary patterns for biological entities or delimiting species based on trees (50).
One method for estimating the phylogenetic signal is tree topology comparison using several methods of analysis. For DNA-based matrices, ML and BI are often suggested over MP because they use an explicit model of evolution, have a consistent approach to parameter estimation problems, have lower variance problems, do not violate many of the assumptions of the evolutionary model, and better account for branch length (51). In addition, ML can be more accurate than BI due to possible long branch attraction in trees (52,53). Our results suggest a consistent topology from both approaches (ML and BI) but a weak phylogenetic signal to differentiate Central America A. neivai specimens from those in Colombia.
For anopheline phylogeny, the use of a 460 bp fragment of Cytb usually lacks meaningful phylogenetic signal due to extreme conservation at the protein level and rapid saturation of synonymous positions for reconstructing the phylogeny among eight subgenera in Anopheles (Anopheles, Cellia, Nyssorhynchus, Kerteszia, Stethomyia, Bironellaand Chagasia) (13). However, the use of a fragment of this gene, as with additional loci (RNASer-IG1ND1) in this work, improved the estimation of tree topology in A. neivai.
In addition, transfer RNA is one of the most central and ancient molecules of the cell (54). Theevolution of this molecule supports the hypothesis of accretion of its structural parts where acceptor arm and TØC loop are known to be more ancient than the anticodon, the DHU and the variable loop (55). Because of its importance in cell protein biosynthesis, the tRNA secondary structure is highly conserved at DHU and TØC loops (56). However, polymorphisms at stems caused by a switch from a Watson-Crick base pairing (A-U, or G-C) to non Watson-Crick pairing (A-C or G-U), could affect the fitness of individuals (57) . Our results from tRNASer secondary structure revealed there is a switch from G-C (variant B) to G-U (variant A) at the DHU stem. In evolving lineages, the fixation at population level of intermediates from this switch phenomenon is rare (56). However, the switch from Watson-Crick to non-Watson-Crick base pairing can compromise the development of bristle and decrease fecundity in other insects as reported in Drosophila (58).
The use of this region in anophelines or other culicids is scarce, despite the efficacy of tRNASer for resolving cryptic species in insects such as sandflies Lutzomyia pia and L. tihuilensis (Diptera: Psychodidae) (59) and differentiation among satyr butterflies (29). From our results, the polymorphism in A. neivai from a single transition occurring at the DHU stem cannot be considered different biological entities but instead a putative ancestral variant (B) and a derived intermediate form (variant A), as variant B shares the same Watson-Crick pairing at DHU stem with variant C from A. pholidotus.
In insect mitogenomes, a small conserved region between tRNASer and ND1 has been reported previously (35,60,61). For anophelines and other insects the size of this region can vary as result of insertions and deletions. The phylogenetic utility of IG1for resolving species complexes was demonstrated when comparing mutations (insertions) between A. gambiae and A. quadrimaculatus (62). However, the current results suggest that substitution patterns and indels in IG1 make it unsuitable for phylogenetic reconstruction and molecular taxonomy as consequence of length (25 bp) and low polymorphism. In addition, the motif (ATACTAA) for A. neivai and A. pholidotus corresponds to the binding site of the transcription termination peptide (MtTERM), signifying the end of the major strand coding region (63,64) shared among insect mtDNA (63).
Currently, the use of integrative taxonomy is multifaceted, using morphological and genetic evidence for the delimitation of the biological entities and integrating different perspectives, such as molecular systematics, phylogeography, comparative biology, and population genetics (65). From our perspective, the utility of the Cytb-SertRNA-IG1-ND1 region revealed interesting polymorphisms in A. neivai from a wide geographic range from Guatemala, the type locality in Panamá, the Pacific coast and the Urabá gulf in Colombia. However, the variations were not considered adequate to uncover cryptic species. Several variants may be adaptive and appear to be distributed in relation to altitude (Chiquimula, Guatemala) and geographic distribution (comparing Panamá, Guatemala and Colombia). The significance of these genetic variants requires further verification by additional studies on biting behavior, human affinity, mitogenomics and population dynamics.
We would like to thank Natalia Molina for her help on processing part of the specimens from Litoral de San Juan; Diego Puerta and Jovany Barajas for their collaboration during the collecting trips. Special thanks go to Dr. Paula Marcet, for her help and assistance for sequencing DNA samples.
All authors report no conflict of interest and state their responsibility for the content and writing of this article.
We would like to thank Colciencias (567-Ph.D. grant), Universidad Nacional de Colombia, sede Medellín, and PECET – Universidad de Antioquia, for funding the collecting surveys, reagents and equipment employed in this research.
Corresponding author:
Andrés López-Rubio, Calle 59 A N° 63-20, bloque 16, laboratorio 110, Medellín, Colombia Teléfono: (313) 624 4845
1. Zavortink TJ. Mosquito studies (Diptera, Culicidae) XXIX. A review of the subgenus Kerteszia of Anopheles. Contrib Am Entomol Inst. 1973;9:1-54.
2. Escovar J, González R, Quiñones ML. Anthropophilic biting behaviour of Anopheles (Kerteszia) neivai Howard, Dyar & Knab associated with Fishermen’s activities in a malaria-endemic area in the Colombian Pacific. Mem Inst Oswaldo Cruz. 2013;108:1057-64. https://doi.org/10.1590/0074-0276130256
3. Murillo C, Jaramillo C, Quintero J, Suárez M. Biología de Anopheles (Kerteszia) neivai H., D. & K., 1913 (Diptera: Culicidae) en la costa pacífica de Colombia. IV Estructura etárea y transmisión de malaria. Rev Saúde Pública. 1989;23:363-7. https://doi.org/10.1590/S0034-89101989000500001
4. González R, Carrejo N. Introducción al estudio taxonómico de Anopheles de Colombia: claves y notas de distribución. Cali: Programa Editorial Universidad del Valle; 2009. p. 260.
5. Gutiérrez LA, Naranjo N, Jaramillo LM, Muskus C, Luckhart S, Conn JE, et al . Natural infectivity of Anopheles species from the Pacific and Atlantic Regions of Colombia. Acta Trop. 2008; 107:99-105. https://doi.org/10.1016/j.actatropica.2008.04.019
6. Murillo C, Astaiza R, Fajardo P. Biología de Anopheles (Kerteszia) neivai H., D. & K., 1913 (Diptera:Culicidae) en la Costa Pacífica de Colombia. III Medidas de luminosidad y el comportamiento de picadura. Rev Saúde Pública. 1988;22:109-12. https://doi.org/10.1590/S0034-89101988000200006
7. Astaiza R, Murillo C, Fajardo P. Biología de Anopheles (Kerteszia) neivai H., D. & K., 1913 (Diptera: Culicidae) en la Costa Pacífica de Colombia. II Fluctuación de la población adulta. Rev Saúde Pública. 1988;22:101-8. https://doi.org/10.1590/S0034-89101988000200005
8. Collins FH, Paskewitz SM. A review of the use of ribosomal DNA (rDNA) to differentiate among cryptic Anopheles species. Insect Mol Biol. 1996;5:1-9. https://doi.org/10.1111/j.1365-2583.1996.tb00034.x
9. Rosero DA, Jaramillo LM, Gutiérrez LA, Conn JE, Correa MM. Genetic diversity of Anopheles triannulatuss. l. (Diptera: Culicidae) from Northwestern and Southeastern Colombia. Am J Trop Med Hyg. 2012;87:910-20. https://doi.org/10.4269/ajtmh.2012.12-0285
10. Lehr MA, Kilpatrick CW, Wilkerson RC, Conn JE. Cryptic species in the Anopheles (Nyssorhynchus) albitarsis (Diptera: Culicidae) complex: Incongruence between random amplified polymorphic DNA-polymerase chain reaction identification and analysis of mitochondrial DNA COI gene. Ann Entomol Soc Am. 2005;98:908-17. https://doi.org/10.1603/0013-8746(2005)098[0908:CSITAN]2.0.CO;2
11. Montoya-Lerma J, Solarte Y, Giraldo-Calderón GI, Quiñones ML, Ruiz-López F, Wilkerson RC, et al . Malaria vector species in Colombia: A review. Mem Inst Oswaldo Cruz. 2011;106:223-38. https://doi.org/10.1590/S0074-02762011000900028
12. Linton Y-MM. Mosquito Barcoding Initiative. The first barcode release paper. Third International Barcode of Life Conference. México: Consortium for the Barcoding of LifeCBOL. – 2009. Accessed: January 15 th , 2017. Available from: https://vimeo.com/8996184
13. Krzywinski J, Wilkerson RC, Besansky NJ. Evolution of mitochondrial and ribosomal gene sequences in Anophelinae (Diptera: Culicidae): Implications for phylogeny reconstruction.
Mol Phylogenet Evol. 2001;18:479-87. https://doi.org/10.1006/mpev.2000.0894
14. Freitas LA, Russo CA, Voloch CM, Mutaquiha OC, Marques LP, Schrago CG. Diversification of the genus Anopheles and a neotropical clade from the late Cretaceous. PLoS One. 2015;10:e0134462. https://doi.org/10.1371/journal.pone.0134462
15. Foster PG, Bergo ES, Bourke BP, Oliveira TM, Nagaki SS, Sant’Ana DC, et al . Phylogenetic analysis and DNAbased species confirmation in Anopheles (Nyssorhynchus). PLoS One. 2013;8:e54063. https://doi.org/10.1371/journal.pone.0054063
16. Ruiz-López F, Wilkerson RC, Conn JE, McKeon SN, Levin DM, Quiñones ML, et al . DNA barcoding reveals both known and novel taxa in the Albitarsis group (Anopheles: Nyssorhynchus) of Neotropical malaria vectors. Parasit Vectors. 2012;5:44. https://doi.org/10.1186/1756-3305-5-44
17. Moreno M, Bickersmith S, Harlow W, Hildebrandt J, McKeon SN, Silva-do-Nascimento TF, et al . Phylogeography of the neotropical Anopheles triannulatus complex (Diptera: Culicidae) supports deep structure and complex patterns. Parasit Vectors. 2013;6:47. https://doi.org/10.1186/1756-3305-6-47
18. Hoy AM. Insect Molecular Genetics. An introduction to principles and applications. 3rd edition. Boston, MA: Academic Press; 2013. p. 364.
19. Rubinoff D, Holland BS. Between two extremes: Mitochondrial DNA is neither the panacea nor the nemesis of phylogenetic and taxonomic inference. Syst Biol. 2005;54: 952-61. https://doi.org/10.1080/10635150500234674
20. Hurst GD, Jiggins FM. Problems with mitochondrial DNA as a marker in population, phylogeographic and phylogenetic studies: The effects of inherited symbionts. Proc R Soc B Biol Sci. 2005;272:1525-34. https://doi.org/10.1098/rspb.2005.3056
21. Norris LC, Norris DE. Phylogeny of anopheline (Diptera: Culicidae) species in southern Africa, based on nuclear and mitochondrial genes. J Vector Ecol. 2015;40:16-27. https://doi.org/10.1111/jvec.12128
22. Harbach RE. The phylogeny and classification of Anopheles. In: Manguin S, editor. Anopheles mosquitoes - New insights into malaria vectors. Rijeka: InTech; 2013.
23. López-Rubio A, Suaza-Vasco J, Marcet PL, Ruiz-Molina N, Cáceres L, Porter C, Uribe S. Use of DNA barcoding to distinguish the malaria vector Anopheles neivai in Colombia. Zootaxa. 2016;4175:377-89. https://doi.org/10.11646/zootaxa.4175.4.7
24. Danforth BN, Lin CP, Fang J. How do insect nuclear ribosomal genes compare to protein-coding genes in phylogenetic utility and nucleotide substitution patterns? Syst Entomol. 2005;30:549-62. https://doi.org/10.1111/j.13653113.2005.00305.x
25. Krzywinski J, Wilkerson RC, Besansky NJ. Toward understanding Anophelinae (Diptera, Culicidae) phylogeny: Insights from nuclear single-copy genes and the weight of evidence. Syst Biol. 2001;50:540-56. https://doi.org/10.1080/10635150119931
26. Vivero RJ, Contreras-Gutiérrez MA, Bejarano EE. Análisis de la estructura primaria y secundaria del ARN de transferencia mitocondrial para serina en siete especies de Lutzomyia. Biomédica. 2007;27:429-38. https://doi.org/10.7705/biomedica.v27i3.205
27. Yona AH, Bloom-Ackermann Z, Frumkin I, Hanson-Smith V, Charpak-Amikam Y, Feng Q, et al . tRNA genes rapidly change in evolution to meet novel translational demands. Elife. 2013;2:e01339. https://doi.org/10.7554/eLife.01339.001
28. Kumazawa Y, Nishida M. Sequence evolution of mitochondrial tRNA genes and deep-branch animal phylogenetics. J Mol Evol. 1993;37:380-98. https://doi.org/10.1007/BF00178868
29. Marín MA, López A, Uribe SI. Interspecific variation in mitochondrial serine transfer RNA (UCN) in Euptychiina butterflies (Lepidoptera: Satyrinae): Structure and alignment. Mitochondrial DNA. 2012;23:208-15. https://doi.org/10.3109/19401736.2012.668895
30. Keller A, Förster F, Müller T, Dandekar T, Schultz J, Wolf M. Including RNA secondary structures improves accuracy and robustness in reconstruction of phylogenetic trees. Biol Direct. 2010;5:4. https://doi.org/10.1186/1745-6150-5-4
31. Pecor J, Gaffigan T. Collecting, rearing, preserving, mounting and shipping techniques for mosquitoes. Walter Reed Biosystematics Unit. 1997. Accessed: January 4th, 2017. Available from: http://www.wrbu.org/about/techniques.html
32. González R, Carrejo N. Introducción al estudio taxonómico de Anopheles de Colombia: claves y notas de distribución. Cali: Programa Editorial Universidad del Valle; 2009. p. 260.
33. Harrison BA, Ruiz-López F, Falero GC, Savage HM, Pecor JE, Wilkerson RC. Anopheles (Kerteszia) lepidotus (Diptera: Culicidae), not the malaria vector we thought it was: Revised male and female morphology; larva, pupa, and male genitalia characters; and molecular verification. Zootaxa. 2012;3218:1-17.
34. Uribe SI, Porter CH, Vélez ID. Amplificación y obtención de secuencias de rRNA mitocondrial en Lutzomyia spp. (Diptera: Psychodidae), vectores de leishmaniosis. Rev Colomb Entomol. 1998;23:177-85.
35. Ready PD, Day JC, de Souza A, Rangel EF, Davies CR. Mitochondrial DNA characterization of populations of Lutzomyia whitmani (Diptera: Psychodidae) incriminated in the peri-domestic and silvatic transmission of Leishmania species in Brazil. Bull Entomol Res. 1997;87:187-95. https://doi.org/10.1017/S0007485300027346
36. Simon C, Frati F, Beckenbach A, Crespi B, Liu H, Flook P. Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann Entomol Soc Am.1994;87:651-701. https://doi.org/10.1093/aesa/87.6.651
37. Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, et al . Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647-9. https://doi.org/10.1093/bioinformatics/bts199
38. Johnson M, Zaretskaya I, Raytselis Y, Merezhuk Y, McGinnis S, Madden TL. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008;36:W5-9. https://doi.org/10.1093%2Fnar%2Fgkn201
39. Hlaing T, Tun-Lin W, Somboon P, Socheat D, Setha T, Min S, et al . Mitochondrial pseudogenes in the nuclear genome of Aedes aegypti mosquitoes: Implications for past and future population genetic studies. BMC Genet. 2009;10:11. https://doi.org/10.1186/1471-2156-10-11
40. Larkin MA, Blackshields G, Brown NP, Chenna R, Mcgettigan PA, McWilliam H, et al . Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947-8. https://doi.org/10.1093/bioinformatics/btm404
41. Librado P, Rozas J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009; 25:1451-2. https://doi.org/10.1093/bioinformatics/btp187
42. Lorenz R, Bernhart SH, Höner-zuSiederdissen C, Tafer H, Flamm C, Stadler PF, et al . ViennaRNA Package 2.0. Algorithms Mol Biol. 2011;6:26. https://doi.org/10.1186/17487188-6-26
43. Mathews DH, Disney MD, Childs JL, Schroeder SJ, Zuker M, Turner DH. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc Natl Acad Sci USA. 2004;101:7287-92. https://doi.org/10.1073/pnas.0401799101
44. Kumar S, Stecher G, Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870-4. https://doi.org/10.1093/molbev/msw054
45. Tavaré S. Some probabilistic and statistical problems in the analysis of DNA sequences. Lect Math Life Sci. 1986;17:57-86.
46. Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307-21. https://doi.org/10.1093/sysbio/syq010
47. Yule GU. A mathematical theory of evolution based on the conclusions of Dr. J.C. Willis, F.R.S. J R Stat Soc. 1925;88:433-6. https://doi.org/10.1098/rstb.1925.0002
48. Bouckaert RR, Heled J, Kühnert D, Vaughan T, Wu C-H, Xie D, et al . BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Comput Biol. 2014;10:e1003537. https://doi.org/10.1371/journal.pcbi.1003537
49. Münkemüller T, Lavergne S, Bzeznik B, Dray S, Jombart T, Schiffers K, et al . How to measure and test phylogenetic signal. Methods Ecol Evol. 2012;3:743-56. https://doi.org/10.1111/j.2041-210X.2012.00196.x
50. Blomberg SP, Garland T, Ives AR. Testing for phylogenetic signal in comparative data: Behavioural traits are more labile. Evolution (N Y). 2003;57:717-45. https://doi.org/10.1111/j.0014-3820.2003.tb00285.x
51. Felsenstein J. Inferring Phylogenies. Sunderland: Sinauer Associates; 2004. p. 664.
52. Kolaczkowski B, Thornton JW. Long-branch attraction bias and inconsistency in Bayesian phylogenetics. PLoS One. 2009;4:e7891. https://doi.org10.1371/journal.pone.0007891
53. Susko E. Bayesian long branch attraction bias and corrections. Syst Biol. 2015;64:243-55. https://doi.org10.1093/sysbio/syu099
54. Caetano-Anollés G, Sun FJ. The natural history of transfer RNA and its interactions with the ribosome. Front Genet. 2014;5:1-5. https://doi.org/10.3389/fgene.2014.00127
55. Sun FJ, Caetano-Anollés G. The origin and evolution of tRNA inferred from phylogenetic analysis of structure. J Mol Evol. 2008;66:21-35. https://doi.org/10.1007/s00239-007-9050-8
56. Meer M V, Kondrashov AS, Artzy-Randrup Y, Kondrashov FA. Compensatory evolution in mitochondrial tRNAs navigates valleys of low fitness. Nature. 2010;464:279-82. https://doi.org/10.1038/nature08691
57. Zhang J, Ferré-D’Amaré A. The tRNA elbow in structure, recognition and evolution. Life. 2016;6:3. https://doi.org/10.3390/life6010003
58. Meiklejohn CD, Holmbeck MA, Siddiq MA, Abt DN, Rand DM, Montooth KL. An incompatibility between a mitochondrial tRNA and its nuclear-encoded trnasynthetase compromises development and fitness in Drosophila. PLoS Genet. 2013;9:e1003238. https://doi.org/10.1371/journal.pgen.1003238
59. Pérez-Doria A, Bejarano EE, Sierra D, Vélez ID. Molecular evidence confirms the taxonomic separation of Lutzomyia tihuiliensis from Lutzomyia pia (Diptera: Psychodidae) and the usefulness of pleural pigmentation patterns in species identification. J Med Entomol. 2008;45:653-9. https://doi.org/10.1093/jmedent/45.4.653
60. Dotson EM, Beard CB. Sequence and organization of the mitochondrial genome of the Chagas disease vector, Triatoma dimidiata. Insect Mol Biol. 2001;10:205-15. https://doi.org/10.1046/j.1365-2583.2001.00258.x
61. Kim I, Lee EM, Seol KY, Yun EY, Lee YB, Hwang JS, et al. The mitochondrial genome of the Korean hairstreak, Coreana raphaelis (Lepidoptera: Lycaenidae). Insect Mol Biol. 2006;15:217-25. https://doi.org/10.1111/j.1365-2583.2006.00630.x
62. Beard CB, Hamm DM, Collins FH. The mitochondrial genome of the mosquito Anopheles gambiae: DNA sequence, genome organization, and comparisons with mitochondrial sequences of other insects. Insect Mol Biol. 1993;2:103-24. https://doi.org/10.1111/j.1365-2583.1993.tb00131.x
63. Cameron SL, Whiting MF. The complete mitochondrial genome of the tobacco hornworm, Manduca sexta, (Insecta: Lepidoptera: Sphingidae), and an examination of mitochondrial gene variability within butterflies and moths. Gene. 2008;408:112-23. https://doi.org/10.1016/j.gene.2007.10.023
64. Sheffield NC, Song H, Cameron SL, Whiting MF.A comparative analysis of mitochondrial genomes in
coleoptera (Arthropoda: Insecta) and genome descriptions of six new beetles. Mol Biol Evol. 2008;25:2499-509. https://doi.org/10.1093/molbev/msn198