ARTÍCULO ORIGINAL
1Escuela de Ciencias de la Salud, Universidad Pontificia Bolivariana, Medellín, Colombia
2Grupo de Biología Estructural y Proteómica Group, Universidad Pontificia Bolivariana, Medellín, Colombia
3Fundación Instituto Neurológico de Colombia (INDEC), Medellín, Colombia
4Systems Proteomics Center, School of Medicine, The University of North Carolina at Chapel Hill, Chapel Hill, USA
5Department of Cell and Developmental Biology, School of Medicine, The University of North Carolina at Chapel Hill, Chapel Hill, USA
6Program in Molecular Biology and Biotechnology, School of Medicine, The University of North Carolina at Chapel Hill, Chapel Hill, USA
* These authors contributed equally to this work
Authors’ contributions:
Luz E. Botero, Juan C. Suárez-Escudero, Andrés E. Toro, Alber J. Patiño, Guillermo Salazar, Juan C. Rodríguez and Gustavo A.
Alarcón prepared and designed the research project, made home visits to the patients and control individuals, applied epidemiological surveys, collected data and laboratory samples.
Juan C. Suárez-Escudero, Andrés E. Toro, Guillermo Salazar, Alber J. Patiño and Juan C. Rodríguez made clinical evaluations of all individuals in the study.
Gustavo A. Alarcón and Luz E. Botero performed the initial preparation of the samples for laboratory analysis.
Ana Corcimaru, Cristina Osorio and Joseph S. Y. Jeong prepared the samples and performed the genotyping.
All co-authors analyzed the results, and contributed in drafting the manuscript.
Oscar Alzate coordinated the project, helped with data analysis, and finalized the manuscript.
All authors approved the final version of the manuscript.
Recibido: 13/10/11; aceptado:08/03/12
Introduction. Alzheimer’s disease is a multifactorial disease affecting approximately twenty million people worldwide. Numerous variables are associated with increased risk of developing this severe neurological disorder. Among the risk factors, diabetes mellitus, and the ε4 isoform of the APOE gene have been amply demonstrated as increasing the risk of developing this disease.
Objective. To determine if a correlation exists between APOE genotype, diabetes mellitus and Alzheimer’s disease.
Materials and methods. Clinical studies were carried out by surveying the clinical histories in a group of patients in the province of Antioquia, Colombia. Forty-three Alzheimer’s patients were compared with 43 control subjects, paired by age and gender. Commercially available methods were used to determine whether the patients had diabetes, and restriction enzyme-based genotyping was used to determine the APOE genotypes.
Results. The most common non-neurological comorbidities were: arterial hypertension, acute myocardial infarction, chronic obstructive pulmonary disease and hypothyroidism. From the many variables investigated, two were conclusive: (1) the presence of Alzheimer’s disease was higher in patients with diabetes mellitus, and (2) no correlation between late-onset sporadic Alzheimer’s disease and APOE was found in the target population.
Conclusions. To detect any association with the APOE genotype, a study involving much a larger population samples must be undertaken.
Keywords: Alzheimer’s disease, diabetes mellitus, apolipoprotein E, dementia; pulmonary disease, chronic obstructive.
Diabetes mellitus en pacientes con enfermedad de Alzheimer: descripción clínica y correlación con el genotipo APOE en una muestra de población del departamento de Antioquia, Colombia
Introducción. La enfermedad de Alzheimer es compleja y afecta, aproximadamente, a 20 millones de personas en todo el mundo. Muchas variables parecen aumentar el riesgo de desarrollar esta alteración neurológica. Entre los factores de riesgo, se ha demostrado ampliamente que la diabetes mellitus y la isoforma ε4 del gen APOE tienen incidencia positiva en el desarrollo de la enfermedad.
Se reporta un estudio en el cual se investigó la posible correlación entre APOE, diabetes mellitus y la enfermedad de Alzheimer, en un grupo específico de pacientes del departamento de Antioquia, Colombia.
Objetivo. Determinar si existe una correlación entre APOE, diabetes mellitus y la enfermedad de Alzheimer, en un grupo de pacientes de Antioquia, Colombia.
Materiales y métodos. Se buscaron y analizaron las historias clínicas de los pacientes con diagnóstico de enfermedad de Alzheimer. Se seleccionaron aquellos que cumplían los criterios de inclusión. Se utilizaron métodos comercialmente disponibles para confirmar la presencia de diabetes mellitus. La genotipificación de APOE se hizo con un método basado en la PCR y la digestión con enzimas de restricción, en muestras de todos los participantes en el estudio.
Resultados. En este estudio se analizan 43 casos de enfermedad de Alzheimer y 43 individuos sanos controles, pareados por edad y sexo. Las enfermedades concomitantes no neurológicas más comunes fueron: hipertensión arterial, infarto agudo del miocardio, enfermedad pulmonar obstructiva crónica e hipotiroidismo.
Conclusiones. De las diferentes variables investigadas, dos arrojaron resultados concluyentes: i) la presencia de la enfermedad de Alzheimer es más frecuente en pacientes con diabetes mellitus, y 2) no se encontró correlación entre la enfermedad de Alzheimer de inicio tardío esporádico y el genotipo de APOE. Es importante indicar que debe llevarse a cabo un estudio con un tamaño de población mayor, para determinar cualquier posible correlación o inferencia con el genotipo de APOE.
Palabras clave: enfermedad de Alzheimer, diabetes mellitus, apolipoproteína E, demencia, enfermedad pulmonar obstructiva crónica.
Alzheimer’s disease (AD) is the most common type of dementia affecting memory, problem solving, learning, behavior and the ability to perform everyday activities. It is a syndrome that can be caused by a number of progressive disorders, most of which are currently the subject of intensive research. In the 2010 World Alzheimer Report (http://www.alz.co.uk/research/files/WorldAlzheimer Report2010Executive Summary.pdf), Alzheimer’s Disease International estimated that approximately 36 million people are living with dementia worldwide. Based on this report, nearly two-thirds of the affected population live in low- and middle-income countries, where the “sharpest increases in affected patients are expected to occur”. People with AD, their families and friends are affected on personal, emotional, financial and social levels. Dementia has already affected significantly every health care system in the world (1,2).
AD accounts for 50-70% of dementias (3). AD patients are mainly characterized by a progressive decline in cognitive function concomitant with the appearance of abnormal deposits of the b-amyloid peptide (Ab) called amyloid plaques (4), and neurofibrillary tangles (NFT) composed of aggregates of the protein tau. Other characteristics include synaptic dysfunction (5), decreased cerebral blood flow to the posterior portion of the left cingulate cortex (6), and cerebral endothelial dysfunction (7). Combinations of these and a variety of other factors may lead to neurodegeneration (8). Clinically, AD is defined by the presence of multiple cognitive alterations--characteristically amnesia and one or more of the following disturbances: aphasia, apraxia, agnosia, and alterations of normal executive functioning with progressive neurologic decline (9).
Until now, definitive diagnosis of AD has been by postmortem neuropathological analysis, based on the following observations: diffuse atrophy (mainly in the hippocampus, Sommer’s CA1 sector), decreased number of neurons in the basal nucleus of Meynert, hyperphosphorylated neurofibrillary tangles (hyperphosphorylated tau protein), senile plaques, granulovacuoles (tubulin) and Hirano bodies (actin) (10). These histopathological features are commonly found in the hippocampus, temporal cortex, and basal nucleus of Meynert (lateral septum). In a normally-aging brain, low levels of these alterations are expected, compared to the large amounts in an AD-affected brain (10,11).
The etiology of AD has not yet been clearly established (12). Both heredity and environmental conditions may play important roles in the onset and progression of AD, as suggested by the existence of different clinical presentations (i.e., early-onset AD (EOAD, divided into early-onset familial or eFAD, and early-onset sporadic) and late-onset AD (LOAD, also divided into late-onset familial and late-onset sporadic)) (12,13). Age is the most significant risk factor for AD (14). Autosomal dominant EOAD (younger than 65 years of age) has been associated with point mutations on the APP (amyloid precursor protein) or the PSN1, PSN2 (presenilin) genes. However, this form of the disease accounts for less than 5% of the total number of AD patients (12). The APOE gene, which codes coding for the apolipoprotein E (apoE), has been associated with increased risk of developing LOAD -the most common form of the disease (15).
Diabetes mellitus (DM) is characterized by chronic systemic hyperglycemia in which defects in insulin secretion, peripheral resistance to insulin and increased production of glucose alter the metabolism of carbohydrates, fats and proteins (16). Chronic hyperglycemia in diabetes has been associated with long-term damage of the eyes, kidneys, heart, nervous system and blood vessels (17,18). Four main categories of DM have been identified: Type 1 DM (DM1), Type 2 DM (DM2), other specific types and gestational diabetes (16).
Interestingly, the prevalence of DM1 and DM2 is increasing worldwide (19). DM increases with age, and, in individuals older than 60 years of age, the prevalence of DM has been estimated to be as high as 21% (20). DM prevalence is similar among men and women throughout most age ranges (10.5% and 8.8% in individuals >20 years of age) except for people older than sixty years of age, in which men present a slightly greater prevalence (21). More than 15 million people have DM in Latin America, a number that is expected to increase (22). The diagnosis of DM2 is usually late and often results from the emergence of various complications including retinopathy (ranging from 16-21%), nephropathy (12-23%) and neuropathy (25-40%), with the result that about 30-50% of people suffering from DM2 are unaware of their condition (23).
A recent estimate suggests that diabetes is the fifth leading cause of death worldwide and is responsible for almost 3 million deaths annually (21). DM is included in the list of chronic non-transmissible diseases. The magnitude of this WHO epidemiological category is such that it accounts for 60% of all deaths worldwide, surpassing by a high margin the group of infectious diseases, including HIV/AIDS, TB and malaria.
DM is another risk for AD (24). The relationship between DM and AD has been found to be particularly strong in patients with the ε4 allele of APOE (APOE4) (25), and DM has been proposed to be associated with sporadic and familial LOAD (26-28).
DM2 and the APOE4 allele have been found to be associated with an increased amount of amyloid plaques in the hippocampus, increased amount of NFT and increased cerebral amyloid angiopathy (8,29,30). ApoE is a circulating lipoprotein, which, by binding the low density lipoprotein (LDL) receptor, promotes the intracellular metabolism of lipids (31-33). This protein is vital for the normal metabolism of triglyceride-rich lipoproteins (32). The APOE gene is polymorphic, with three common alleles (ε2, ε3, and ε4); these are linked to different lipid metabolic profiles, but not showing a particular distribution in DM2 (34).
The APOE4 genotype increases the risk of developing AD (15). ApoE is important for maintenance and repair of neurons and apoE3 is more efficient than apoE4 in this regard (33). Histopathological data indicate that the expression of the insulin-degrading enzyme in the hippocampus is substantially reduced relative to controls, in particular in AD patients with the APOE4 allele (35).
The genotype displaying the highest risk for developing sporadic LOAD is APOE4/4 (15). The APOE3/4 genotype is associated with a medium-to-high risk for AD. APOE3/3 genotype has a risk factor equal to the general population. APOE2/3 genotype is correlated with a lower risk for developing AD. Those with APOE2/4, have a risk equivalent to APOE3/3, i.e., similar to the rest of the population (36). In the general population the frequency of the ε3 allele is 78% and of the ε4 allele is 13%. In patients with LOAD, the ε4 allele frequency is 52%. In fact, homozygous APOE4/4 people present almost 8 times higher risk of developing AD compared to homozygous APOE3/3 people; although homozygousity for ε4 does not necessarily result in developing the disease (15,36). Among individuals from families with LOAD without copies of ε4, only 20% develop AD at around 75 years of age, compared to 90% of individuals with two copies of the ε4 allele (37).
In Colombia, the frequencies of the ε2, ε3, and ε4 alleles are similar to those reported in other countries. In a study conducted in 2006 in Bogotá involving residents between 18 and 106 years of age, 75% of the population were found with the APOE3/3 genotype, 15% were of the APOE3/4 genotype, 8.2% were APOE2/3, 1.4% were of the APOE2/4 genotype, and 0.7% of the population were APOE2/2 or APOE4/4 (38). Similar results were found by another study conducted in 2007 involving 1,567 individuals from the Central and East parts of the country (39).
The glycemic dysregulation found in DM has been associated with brain structural damage, including damage in large and small vessels (40). Hyperglycemia can affect neural viability by increasing oxidative stress and formation of advanced glycation end-products (AGEs) (41). It is now considered that the AGEs play an important role in the pathophysiology of various neurological diseases via several mechanisms: (1) by modifying extracellular structural proteins; (2) by triggering intracellular processes that bind to extracellular receptors activating signal cascades; and (3) by modifying intracellular proteins. In diabetic patients, the accumulation of AGEs leads to endothelial dysfunction among other important structural changes (41-43).
Hyperglycemia also has a direct effect on neural calcium balance in the hippocampus resulting in cell damage. It may also produce hyperosmolarity and increase in vasopressin levels, eventually leading to hypothalamic neuronal degeneration (40,44). Altered glucose metabolism may result in decreased acetylcholine production, synaptic failure, and cholinergic neuron degeneration in the hippocampus (45,46). The appearance of atherosclerosis in cerebral arteries is also associated with DM, resulting in decreased cerebral blood flow and a disturbance in vascular reactivity causing hypoxic and ischemic states, which are important pathological conditions that can cause multiple vascular dementias (6,47,48). Biesselsetal. (49) concluded that DMincreases the riskof atherosclerosisand stroke, (i.e., something that produces acerebralvasculardisease, mediatedby glucosetoxicityandmicrovascular abnormalities), both of which lead toacceleratedbrainaging (49,50).
The accelerated formation of AGEs has been found to correlate with an increase in Ab deposition, greater number of plaques, neurofibrillary tangles and elevated tau protein in the hippocampus and the cerebral cortex. Furthermore, accelerated AGEs formation is closely related to increase in oxidative stress (51). Insulin and glucose dysregulation can also alter amyloid metabolism by changing the insulin/insulin receptor interaction (52) and by the formation of AGEs (53).
DM is another risk for AD, although the molecular pathways connecting DM and AD are unknown (24). The fourth and fifth stages (known as TheHonolulu Asia Aging Study) of the so-called “Honolulu Heart Program” were designed to investigate risk factors associated with neurodegenerative disorders and aging. This research program also presented substantial evidence that diabetes is a major risk factor for the development of both vascular dementia and AD (25). It was also found that the association was particularly stronger in APOE ε4 carriers (25). Similar results were also obtained by the “The Cardiovascular Health Study Cognition”, a cohort study carried out from 1992 to 2000 (54).
Based on these previous publications in which DM2 and the APOE genotype have been found as risk factors for AD, the current study was designed to determine if a correlation was present among these variables, in a cohort of patients from the Aburrá Valley (province of Antioquia, Colombia), who had been diagnosed with AD. Despite using a small population (43 cases and 43 controls), a statistically significant correlation was found between DM2 and AD. All patients and controls, selected at random for this study, were either APOE3/3 or APOE 2/3, with no ε4 alleles present. Clearly, an additional future study including a larger randomized sample is necessary before a correlation can be inferred between the APOE genotype and AD.
Materials and methods
The study design was a descriptive case-control study. The study population was composed of individuals residing in the towns of Medellín, Envigado, Itagüí, La Estrella, Bello, and Copacabana (Antioquia Province, Colombia). The population sample (n=86) was divided into two groups: individuals with diagnosis of AD who met the DSM-IVR criteria for AD diagnosis (n=43) and control subjects who did not meet a single DSM-IVR criteria for AD (n=43).
AD patients were selected according to the following characteristics: age ≥ 65 years of age regardless of sex, diagnosed with sporadic LOAD by a neurologist, meeting the DSM-IVR clinical criteria for its diagnosis, and having no relatives with an AD diagnosis (thus selecting only for sporadic LOAD) or any other type of dementia. All patients and family members (guardian or caretaker) as well as the control group population participated voluntarily and were recruited for this study following the regulations of the ethics committee of the “Universidad Pontificia Bolivariana” and from the ethics and research committee of the Instituto Neurológico de Colombia (INDEC).
In order to validate the diagnosis of AD, the following pathological conditions were ruled out: stroke, Parkinson’s disease, Huntington’s chorea, chronic subdural hematoma, normal-pressure hydrocephalus, cerebral neoplasm, depressive pseudo dementia, hypothyroidism, folic acid deficiency, vitamin B12 deficiency, neurosyphilis, HIV/AIDS, TB, Wilson’s disease, cysticercosis, uremic encephalopathy, hepatic encephalopathy, autoimmune processes, and chronic sleep apnea. This was done by analyzing the medical history of the patients (who have a long term follow-up from the INDEC) and retrospective corroboration with their families at the time of the home visit.
The control group (healthy individuals) consisted of persons older than 65 years of age from both genders, without medical history of dementia syndrome (9) or any other neurodegenerative disease. These individuals did not meet any of the DSM-IVR criteria for AD, had no history of stroke, head injury, or neurosurgery at any point in their lives, and had no family history of AD.
In addition, all individuals included in the study were evaluated for DM. INDEC patients and control subjects were evaluated according to their clinical history and by validation of the condition with laboratory tests using methods commercially available. Individuals who were on steroids over a 3-month period previous to this study, or who had any glycemic alterations resulting from steroids intake, were not included in this study.
Selection of individuals
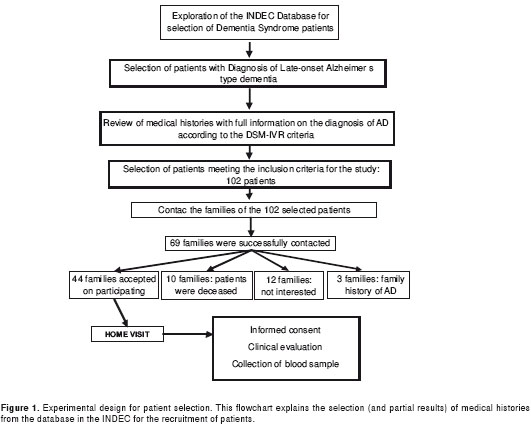
The inclusion of subjects in the study was made on the basis of a database from the INDEC that included 779 medical records of patients with diagnosis of dementia syndrome in the Neurology Outpatient service, from January 2008 to July 2009. Four hundred seventeen patients, whose medical histories documented the clinical diagnosis of LOAD, were pre-selected (ICD-10 code: G30.1). From these, 190 were selected through meticulous and detailed reading of the patient’s diagnostic records. Of these, 102 met the selection criteria. Following strict regulations of the INDEC protecting the privacy of the patients and their families, families of 69 patients were contacted, and 44 agreed to participate in the study-which included a home visit with each family. This experimental design is presented in figure 1.
The privacy of the participants was protected by dividing the research team into two groups. One group consisted of physicians and nurses, who interacted with the patients and their families, whereas the other team consisted of scientists performing molecular studies. At no point during this investigation did the groups exchange personal information. During the home visit, a physician from the INDEC provided a physical examination including a neurological exam to corroborate the data recorded on the medical histories (internal validity) and filled out a form with the patient’s clinic-epidemiological survey. This step ascertained that the patient met the DSM-IVR diagnostic criteria for AD as already stated by the medical history. This meeting was followed by blood sampling (as described below). One patient was withdrawn from the group because it was confirmed that there was AD history in the family.
During the home visits healthy individuals were recruited from the community and were paired with AD patients according to gender and age (± 5 years). Control subjects met the pre-established inclusion and exclusion criteria and were visited by the group of researchers to their respective domiciles. At this point the clinic-epidemiological survey form was filled out, followed by blood sampling for laboratory testing.
Clinic-epidemiological survey
Since each medical history documented the diagnosis of AD, a complete anamnesis was conducted in order to confirm the information found in the medical history from the database. Therefore, each individual underwent a physical examination to determine the individual’s arterial pressure, weight, height, abdominal circumference and body mass index (BMI). Individuals also underwent a complete neurological examination, verifying the health status of both AD patients and control subjects and whether they met the DSM-IVR criteria for AD.
Blood sampling, laboratory testing and DNA extraction
Approximately 10 mL of peripheral blood were taken from each individual by venipuncture (it was made sure that each patient had been fasting), using vacuum tubes (BD Vacutainer, Becton Dickinson and Co., BD&C, Franklin Lakes, NJ, USA), and 21G x 1½” needles (BD VacutainerPrecisionGlide, Multiple Sample Needle, BD&C). A portion of the blood (5mL) was collected in tubes with EDTA (BD Vacutainer K2 EDTA, Plus Blood Collection Tubes, BD&C) for extracting leukocytes which was the source of DNA for APOE genotyping. The rest of each sample (5 mL) was collected in reactant- and preservative-free tubes (BD Vacutainer Serum Blood Collection Tubes, BD&C) for blood chemistry analysis. Blood chemistry tests were performed at the clinical laboratory of the “Hospital Universitario Bolivariano” (University Hospital of the Universidad Pontificia Bolivariana). Fasting glucose levels, lipid profiling (total cholesterol, TC; low density lipoprotein, LDL; high density lipoprotein, HDL; very low density lipoprotein, VLDL; and atherogenic index; TC/HDL) were determined using standard procedures.
For DNA extraction, all chemicals were from Bio-Rad (Hercules, California, USA) or Sigma (St. Louis, Missouri, USA). Genomic DNA was extracted from 5 mL of peripheral blood, anti-coagulated in EDTA following the extraction method described by Miller (55) with some modifications. The DNA pellet was allowed to dry and was resuspended in 1 mL of sterile type I DNAase-free H2O and stored at −20ºC until use. For genotyping, DNA samples were quantified and its quality was evaluated using Thermo Scientific NanoDrop 2000c by reading the absorbance of 1 µL of sample at 260nm. The 260/280 absorbance ratio was used to assess the purity of DNA. A ratio of ~1.8 was accepted as “pure” for DNA.
Genotyping of APOE genes was carried out with PCR-based restriction fragment length polymorphism (RFLP) as described by Hixson and Vernier (56) with some modifications. gDNA from human blood samples was amplified with primers (APOE-forward: TAAGCTTGGCACGGCTGTCCAAGGA; APOE-reverse: ACAGAATTCGCCCCGGCCTGGTACACT GCC) to obtain 244bp amplicons. Fifteen microliters of 2X PCR master mix (Fermentas, Glen Burnie, MD, USA), 300 ng of gDNA template, 1.5 µl of 10 µM of each primer and 2 µl of DMSO were mixed to 30 µl reaction. Each reaction mixture was denatured at 94oC for 4 min and 35 cycles of amplification were performed with steps of annealing at 60oC for one min, extension at 72oC for two min and denaturation at 94oC for 30 seconds, followed by an extra extension step at 72oC for three min at the end of the cycles. Ten microliters of PCR product was digested in 30 µl reaction for 3 hours with 5 units of HhaI (New England Biolabs (NEB), Ipswich, MA, USA) with 20 µl of 1X buffer 4 (NEB); 5 µl of each reaction mixture was loaded on an 8% polyacrylamide gel in 1X TBE buffer and run for 3 hours under constant voltage of 10V/1cm. The polyacrylamide gels were stained with ethidium bromide and visualized by UV illumination. As described by Hixson and Vernier (56), HhaI gives different RFLP patterns because of different nucleotide sequences at aminoacid numbers 112 and 158 of ApoE2, ApoE3 and ApoE4 (figure 2).

Ethical considerations
The ethical approval for the execution of this study was obtained from the ethics committee of the “Universidad Pontificia Bolivariana” and from the ethics and research committee of the INDEC.
Access and extraction of data from the INDEC database, as well as the contact with families, was done with previous authorization from the Ethics and Research Committee of the INDEC. Information about the study, including its objectives and methods, was provided by telephone to the families or caretakers of the patients. The home visit was agreed to by the families, or directly with the control subjects. On each visit, a written informed consent was obtained from families or caretakers who signed it voluntarily, and all questions concerning the study were adequately addressed. Protecting the principles of autonomy, respect for the dignity of individuals and beneficence were always a top priority. All the information obtained during the study was confidential. Individuals were assigned with a code in order to protect their identities.
Statistical analysis
The information was obtained and recorded on the clinic-epidemiological survey form, then transferred into a database created with Microsoft Excel®. Central tendency and non-parametric measures were calculated (c2, confidence intervals (CI), relative risk (RR)). Statistically significant data were interpreted with a p-value < 0.05 and their respective confidence intervals.
Results
Forty-three AD cases and 43 control subjects were paired according to their age and gender. The percentage of females in both groups was 53.5%, with an average age for AD cases of 80.9 years (SD=6.7) and the average age for controls 81.7 years (SD=7.5).
The most common non-neurological comorbidities were as follows: arterial hypertension (53.5% for AD cases and 65.1% for control subjects), acute myocardial infarction (11.6% for AD cases and 4.7% for control subjects), chronic obstructive pulmonary disease (18.6% and 4.0%), hypothyroidism (18.6% and 11.6%), smoking (65.1% and 27.9%), alcohol consumption (39.5% and 16.3%), any form of cancer (16.3% and 7.0%), heart failure (11.6% and 7.0%) and dyslipidemia (9.3% and 16.2%). Comorbidities were determined from the medical histories of the population under study and subsequently confirmed with the families of the AD patient and by direct interview with the control subjects. The rest of the clinical diagnosis and history is presented in table 1.
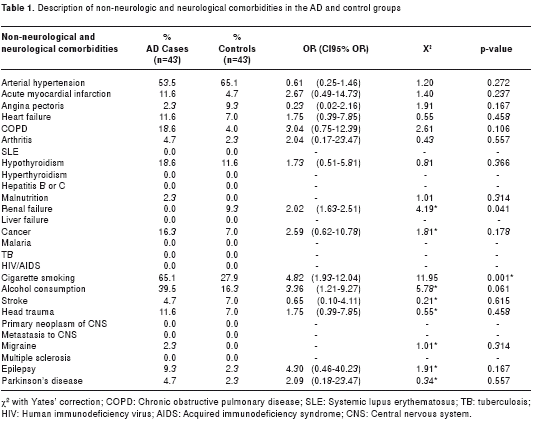
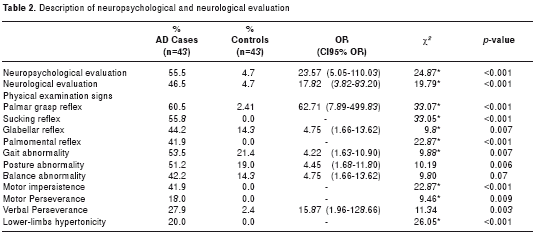
When the non-neurological comorbidities in both groups were compared, significant statistical differences were present for renal failure and smoking (this was the subject status only at the time of analysis; since no long term follow-up was conduted, no conclusions were possible as to whether or not these comorbidities are directly associated with AD or if they represent a causal relationship to AD). The most common neurological comorbidities were head trauma (11.6% and 7.0%) and epilepsy (9.3% and 2.3%), without statistically significant differences. The clinical pathology findings for neuropsychological and neurological evaluation showed statistically significant differences as shown in table 2.
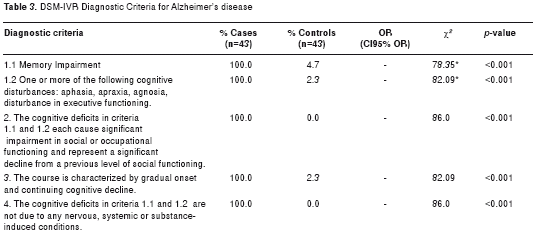
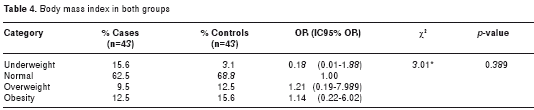
All patients met the DSM-IVR criteria for the diagnosis of AD, displaying in each of them statistically significant differences compared to the control subjects, as shown in table 3. Most individuals in both groups presented normal body mass index (BMI) (62.5% for AD patients and 68.8% for control subjects). Only a quarter of the population was found to have a BMI above normal, 22% in the case group and 28.1% for the control group, as outlined in table 4.
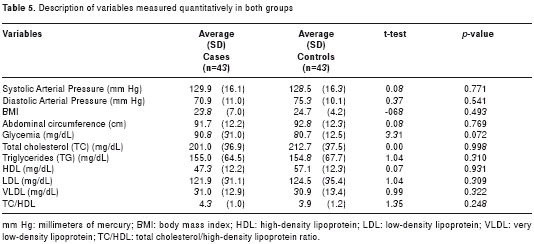
At the time of the home visit, all participants underwent a clinical assessment and blood samples were taken for blood chemistry testing. The values determined for the variables measured are presented in table 5. No significant differences were found in either group. Regarding the presence of alterations in blood glucose levels, eleven of the AD patients (25.6%) and one of the control subjects were diagnosed with DM2 (statistical significance, c2 with Yates’ correction = 9.68, p = 0.002). Diabetic nephropathy, two diabetic retinopathies, and one diabetic foot were diabetic complications found in the AD population. Diabetes was found 14 times more in AD patients than in control subjects (Odds Ratio 14.4, 95% CI 1.7-117.7).
Genotyping studies for APOE indicated that 89% of the population was of the APOE3/3 genotype, while the remaining 11% was of the APOE2/3 genotype. No ε4 allele was found. When evaluating the frequencies of the APOE genotypes in the group of patients and in the group of control subjects, the distribution was comparable to that found in the analysis of the total population (38,39).
Discussion
Many studies have been undertaken to identify potential risk factors for AD. Vascular factors such as arterial hypertension, dyslipidemia, hyperinsulinemia and DM2, obesity, subclinical atherosclerosis and arrhythmias have been associated with high risks of cognitive impairment and dementia (52). Studies in Latin America have also shown that metabolic syndrome doubles the risk of cognitive impairment (57). Kalaria (58) has indicated that the gradual adoption of westernized lifestyle involving excessive caloric intake, unhealthy diet, and decreased physical activity, favors the appearance of conditions such as AD. In the Honolulu-Asia Aging study (25), a significant association was found between diabetes, alone or combined with the APOEε4, with an increase in the incidence of dementia and neuropathological outcomes in a cohort of 2,574 Americans of Japanese origin. This study associated diabetes with total dementia (RR 1.5, 95% CI 1.0-2.2) and with AD (RR 1.8, CI 1.1-2.9). Another study (59,60) found an RR of 1.3 (95% CI 0.9-2.1) for AD. Despite the large differences in the sizes of the sample populations, similarities occur between these reports and the current findings (the strength of a positive association between DM and AD) in which paired analysis found statistically significant correlation between AD and DM (c2 McNemar: 8.18, p= 0.004, with a minimum of 95% CI and OR of 1.90), suggesting that even at a small scale there is a significant correlation. In the most recently published study (24), Ohara et al. showed that subjects with diabetes display a higher incidence of all-cause dementia, AD, and vascular dementia than control subjects with normal glucose tolerance (all-cause dementia: adjusted hazard ratio [HR] = 1.74, 95% CI 1.19-2.53, p=0.004; AD [HR] = 2.05, 95% CI 1.18-3.57, p=0.001).
In the current study, the most common non-neurological comorbidities were as follows: arterial hypertension (53.5% for AD patients and 65.1% for control subjects), acute myocardial infarction (11.6% for AD cases and 4.7% for control subjects), chronic obstructive pulmonary disease (18.6% and 4.0%), hypothyroidism (18.6% and 11.6%), smoking (65.1% and 27.9%), alcohol consumption (39.5% and 16.3%), cancer (16.3% and 7.0%), heart failure (11.6% and 7.0%) and dyslipidemia (9.3% and 16.2%).
Type 1 and type 2 DM have been associated with a progressive decline in cognitive functioning, mainly in elderly patients (49,50,61). Toxic effects of high glucose levels are mediated through an enhanced flux of glucose through the polyol and hexosamine pathways, disturbances of intracellular second messenger pathways, an imbalance in the generation and scavenging of reactive oxygen species, and by advanced glycation of important functional and structural proteins (49). In addition, DM2 is a risk factor for atherosclerosis of the carotid and intracranial arteries, thus increasing the risk of stroke, cognitive decline, and dementia (62,63). In the current study, eleven AD cases (25.6%) and one control subject (2.3%) were given a diagnosis of diabetes--all DM2, representing a statistically-significant difference (p=0.002). However, these results do not imply any causal relationship. The observations are a “one-time-point” observation, and not a follow-up study, and therefore, one cannot conclude from these data that DM leads to an increase in the risk of developing AD. However, this conclusion was reached by Ohara et al. (24) in a 15-year follow-up study.
In the current study sample, smoking with an OR of 4.82 (95% CI 1.93-12.04) suggested it as a risk factor for AD. In general, the literature is inconsistent on this subject. For instance, several studies (64-66) have reported evidence linking smoking more as a protective factor than as a risk factor for AD. The meta-analysis of 19 prospective studies by Anstey, et al. (67), a total of 27,374 participants evaluated for dementia, reported a relative risk of 1.79 (95% CI 1.43-2.23) for AD among smokers and nonsmokers (67). A number of studies reflect the lack of association between these factors (68,69). Given the increasing prevalence of dementia and the high burden of comorbidity, morbidity, and disability, an urgent need is developing to clearly identify whether smoking is a modifiable risk factor for AD.
Among the most prevalent chronic diseases is chronic obstructive pulmonary disease (COPD). In the current study, COPD was found in 18.6% of patients with AD compared with 7% in the control patients. However, the number of patients was not sufficient for concluding an association with COPD. Regarding the presence of cancer, no correlation appears between the presence of cancer and AD (16.3% vs. 7.0% control patients). These data are similar to those in other studies, e.g.,12.15% for AD patients versus 10.11% for control subjects (70).
Finally, the present study is to be considered a preliminary work in which a correlation between APOE genotype, DM, and AD was sought in a specific population. Studies are planned to extend these comparisons to a larger and more diverse population in future.
Acknowledgments
We thankfully acknowledge the anonymous patients, their families and control individuals, who voluntarily participated in the study; and Dr. Carol Parker from the UVic Genome BC Proteomics Centre for careful review of the manuscript.
Conflicts of interest
The authors declare not having any conflict of interest in the research reported here.
Funding
Funding was provided by the Center for Research, Development and Innovation (CIDI, Centro de Investigación para el Desarrollo y la Innovación) of the Universidad Pontificia Bolivariana and by the University of North Carolina Systems Proteomics Center.
Corresponding author: Oscar Alzate, Ph.D., 438A Taylor Hall, Mason Farm Road, Department of Cell and Developmental Biology, CB# 7090, Chapel Hill, NC 27599, USA Phone: (919) 962 3698; fax: (919) 966 1856 alzate@med.unc.edu
References
1. Barnes DE, Yaffe K. The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol. 2011;10:819-28.
2. Wimo A, Winblad B, Jonsson L. The worldwide societal costs of dementia: Estimates for 2009. Alzheimers Dement. 2010;6:98-103.
3. WHO. Atlas: Country resources for neurological disorders. World Health Organization and World Federation of Neurology. Geneva: WHO; 2004. p. 52.
4. Selkoe DJ. Alzheimer’s disease: Genes, proteins, and therapy. Physiol Rev. 2001;81:741-66.
5. Martin JB. Molecular basis of the neurodegenerative disorders. N Engl J Med. 1999;340:1970-80.
6. Huang C, Wahlund LO, Svensson L, Winblad B, Julin P. Cingulate cortex hypoperfusion predicts Alzheimer’s disease in mild cognitive impairment. BMC Neurol. 2002; 2:9.
7. Dede DS, Yavuz B, Yavuz BB, Cankurtaran M, Halil M, Ulger Z, et al. Assessment of endothelial function in Alzheimer’s disease: Is Alzheimer’s disease a vascular disease? J Am Geriatr Soc. 2007;55:1613-7.
8. Dodart JC, Marr RA, Koistinaho M, Gregersen BM, Malkani S, Verma IM, et al. Gene delivery of human apolipoprotein E alters brain A beta burden in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2005;102:1211-6.
9. American Psychiatric Association. Diagnostic and statistical manual of mental disorders. Fourth edition. Washington, DC: APA; 2000. p. 157.
10. Dickson DW. Neuropathological diagnosis of Alzheimer’s disease: A perspective from longitudinal clinicopathological studies. Neurobiol Aging. 1997;18(Suppl.):S21-6.
11. Fagan AM, Holtzman DM. Cerebrospinal fluid biomarkers of Alzheimer’s disease. Biomark Med. 2010;4:51-63.
12. Alzheimer’s Association, Thies W, Bleiler L. Alzheimer’s disease facts and figures. Alzheimers Dement. 2011;7:208-44.
13. Lehtovirta M, Soininen H, Helisalmi S, Mannermaa A, Helkala EL, Hartikainen P, et al. Clinical and neuropsychological characteristics in familial and sporadic Alzheimer’s disease: Relation to apolipoprotein E polymorphism. Neurology. 1996;46:413-9.
14. Yoshitake T, Kiyohara Y, Kato I, Ohmura T, Iwamoto H, Nakayama K, et al. Incidence and risk factors of vascular dementia and Alzheimer’s disease in a defined elderly Japanese population: The Hisayama Study. Neurology. 1995;45:1161-8.
15. Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J,; Salvensen GS, et al. Apolipoprotein E: High-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:1977-81.
16. American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2009;32(Suppl.1):S62-7.
17. Kuzuya T, Nakagawa S, Satoh J, Kanazawa Y, Iwamoto Y, Kobayashi M, et al. Report of the Committee on the classification and diagnostic criteria of diabetes mellitus. Diabetes Res Clin Pract. 2002;55:65-85.
18. Ryden L, Standl E, Bartnik M, van den Berghe G, Betteridge J, de Boer MJ, et al. Guidelines on diabetes, pre-diabetes, and cardiovascular diseases: Executive summary. The Task Force on Diabetes and Cardiovascular Diseases of the European Society of Cardiology (ESC) and of the European Association for the Study of Diabetes (EASD). Eur Heart J. 2007;28:88-136.
19. Danaei G, Finucane MM, Lu Y, Singh GM, Cowan MJ, Paciorek CJ, et al. National, regional, and global trends in fasting plasma glucose and diabetes prevalence since 1980: Systematic analysis of health examination surveys and epidemiological studies with 370 country-years and 2.7 million participants. Lancet. 2011;378:31-40.
20. Lorenzo C, Williams K, Hunt KJ, Haffner SM. The National Cholesterol Education Program - Adult Treatment Panel III, International Diabetes Federation, and World Health Organization definitions of the metabolic syndrome as predictors of incident cardiovascular disease and diabetes. Diabetes Care. 2007;30:8-13.
21. WHO. Diabetes. Geneve: WHO; 2011. Accessed: September 15, 2011. Disponible en: http://www.who.int/mediacentre/factsheets/fs312/en/index.html
22. Aschner P. Diabetes trends in Latin America. Diabetes Metab Res Rev. 2002;18(Suppl.3):S27-31.
23. Alzheimer’s Disease Association. Standards of medical care in diabetes -2011. Diabetes Care. 2011;34(Suppl.1):S11-61.
24. Ohara T, Doi Y, Ninomiya T, Hirakawa Y, Hata J, Iwaki T, et al. Glucose tolerance status and risk of dementia in the community: The Hisayama Study. Neurology. 2011; 77:1126-34.
25. Peila R, Rodríguez BL, Launer LJ. Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: The Honolulu-Asia Aging Study. Diabetes. 2002;51:1256-62.
26. Kuusisto J, Koivisto K, Mykkanen L, Helkala EL, Vanhanen M, Hänninen T, et al. Essential hypertension and cognitive function. The role of hyperinsulinemia. Hypertension. 1993;22:771-9.
27. Leibson CL, Rocca WA, Hanson VA, Cha R, Kokmen E, O’Brien PC, et al. Risk of dementia among persons with diabetes mellitus: A population-based cohort study. Am J Epidemiol. 1997;145:301-8.
28. Ott A, Stolk RP, van Harskamp F, Pols HA, Hofman A, Breteler MM. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology. 1999;53:1937-42.
29. Martins IJ, Hone E, Foster JK, Sunram-Lea SI, Gnjec A, Fuller SJ, et al. Apolipoprotein E, cholesterol metabolism, diabetes, and the convergence of risk factors for Alzheimer’s disease and cardiovascular disease. Mol Psychiatry. 2006;11:721-36.
30. Messier C. Diabetes, Alzheimer’s disease and apolipoprotein genotype. Exp Gerontol. 2003;38:941-6.
31. Mahley RW, Huang Y. Apolipoprotein (apo) E4 and Alzheimer’s disease: Unique conformational and biophysical properties of apoE4 can modulate neuropathology. Acta Neurol Scand Suppl. 2006;185:8-14.
32. Mahley RW, Huang Y, Weisgraber KH. Putting cholesterol in its place: apoE and reverse cholesterol transport. J Clin Invest. 2006;116:1226-9.
33. Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E4: A causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proc Natl Acad Sci U S A. 2006;103:5644-51.
34. Powell DS, Maksoud H, Charge SB, Moffitt JH, Desai M, Da Silva Fihlo RL, et al. Apolipoprotein E genotype, islet amyloid deposition and severity of type 2 diabetes. Diabetes Res Clin Pract. 2003;60:105-10.
35. Edland SD. Insulin-degrading enzyme, apolipoprotein E, and Alzheimer’s disease. J Mol Neurosci. 2004;23:213-7.
36. Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921-3.
37. Levy ML, Cummings JL, Fairbanks LA, Sultzer DL, Small GW. Apolipoprotein E genotype and noncognitive symptoms in Alzheimer’s disease. Biol Psychiatry. 1999;45:422-5.
38. Forero DA, Pinzón J, Arboleda GH, Yunis JJ, Álvarez C, Cataño N, et al. Analysis of common polymorphisms in angiotensin-converting enzyme and apolipoprotein E genes and human longevity in Colombia. Arch Med Res. 2006;37:890-4.
39. Callas N, Poveda E, Baracaldo C, Hernández P, Castillo C, Guerra M. Polimorfismo genético de la apolipoproteína E en un grupo de escolares del centro-oriente colombiano: comparación con las concentraciones plasmáticas de lípidos y apolipoproteínas. Biomédica. 2007;27:526-36.
40. Hernández-Fonseca JP, Rincón J, Pedreanez A, Viera N, Arcaya JL, Carrizo E, et al.Structural and ultrastructural analysis of cerebral cortex, cerebellum, and hypothalamus from diabetic rats. Exp Diabetes Res. 2009;2009:329632.
41. Sasaki N, Fukatsu R, Tsuzuki K, Hayashi Y, Yoshida T, Fujii N, et al. Advanced glycation end products in Alzheimer’s disease and other neurodegenerative diseases. Am J Pathol. 1998;153:1149-55.
42. Brownlee M. Advanced protein glycosylation in diabetes and aging. Annu Rev Med. 1995;46:223-34.
43. Yan SD, Chen X, Schmidt AM, Brett J, Godman G, Zou YS, et al. Glycated tau protein in Alzheimer disease: A mechanism for induction of oxidant stress. Proc Natl Acad Sci U S A. 1994;91:7787-91.
44. Klein JP, Waxman SG. The brain in diabetes: Molecular changes in neurons and their implications for end-organ damage. Lancet Neurol. 2003;2:548-54.
45. Francis PT, Palmer AM, Snape M, Wilcock GK. The cholinergic hypothesis of Alzheimer’s disease: A review of progress. J Neurol Neurosurg Psychiatry. 1999;66:137-47.
46. Nitsch RM. From acetylcholine to amyloid: Neurotrans-mitters and the pathology of Alzheimer’s disease. Neurodegeneration. 1996;5:477-82.
47. Craft S.The role of metabolic disorders in Alzheimer disease and vascular dementia: Two roads converged. Arch Neurol. 2009;66:300-5.
48. Kameyama M, Fushimi H, Udaka F. Diabetes mellitus and cerebral vascular disease. Diabetes Res Clin Pract. 1994;24(Suppl.):S205-8.
49. Biessels GJ, Staekenborg S, Brunner E, Brayne C, Scheltens P. Risk of dementia in diabetes mellitus: A systematic review. Lancet Neurol. 2006;5:64-74.
50. Biessels GJ, Kappelle LJ. Increased risk of Alzheimer’s disease in type II diabetes: Insulin resistance of the brain or insulin-induced amyloid pathology? Biochem Soc Trans. 2005;33:1041-4.
51. Querfurth HW, LaFerla FM. Alzheimer’s disease. N Engl J Med. 2010;362:329-44.
52. Craft S, Watson GS. Insulin and neurodegenerative disease: Shared and specific mechanisms. Lancet Neurol. 2004;3:169-78.
53. Smith MA, Sayre LM, Monnier VM, Perry G. Radical AGEing in Alzheimer’s disease. Trends Neurosci. 1995;18:172-6.
54. Irie F, Fitzpatrick AL, López OL, Kuller LH, Peila R, Newman AB, et al. Enhanced risk for Alzheimer disease in persons with type 2 diabetes and APOE epsilon4: The Cardiovascular Health Study Cognition Study. Arch Neurol. 2008;65:89-93.
55. Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215.
56. Hixson JE, Vernier DT. Restriction isotyping of human apolipoprotein E by gene amplification and cleavage with HhaI. J Lipid Res. 1990;31:545-8.
57. Panza F, Frisardi V, Capurso C, Imbimbo BP, Vendemiale G, Santamato A, et al.Metabolic syndrome and cognitive impairment: Current epidemiology and possible underlying mechanisms. J Alzheimers Dis. 2011;21:691-724.
58. Kalaria RN, Maestre GE, Arizaga R, Friedland RP, Galasko D, Santamato A, et al. Alzheimer’s disease and vascular dementia in developing countries: Prevalence, management, and risk factors. Lancet Neurol. 2008;7:812-26.
59. Qiu C, von Strauss E, Fastbom J, Winblad B, Fratiglioni L. Low blood pressure and risk of dementia in the Kungsholmen project: A 6-year follow-up study. Arch Neurol. 2003;60:223-8.
60. Qiu C, Winblad B, Viitanen M, Fratiglioni L. Pulse pressure and risk of Alzheimer disease in persons aged 75 years and older: A community-based, longitudinal study. Stroke. 2003;34:594-9.
61. Strachan MW, Reynolds RM, Marioni RE, Price JF. Cognitive function, dementia and type 2 diabetes mellitus in the elderly. Nat Rev Endocrinol. 2011;7:108-14.
62. Wannamethee SG, Shaper AG, Lennon L, Morris RW. Metabolic syndrome Vs. Framingham risk score for prediction of coronary heart disease, stroke, and type 2 diabetes mellitus. Arch Intern Med. 2005;165:2644-50.
63. Callahan A, Amarenco P, Goldstein LB, Sillesen H, Messig M, Samsa GP, et al. Risk of stroke and cardiovascular events after ischemic stroke or transient ischemic attack in patients with type 2 diabetes or metabolic syndrome: Secondary analysis of the stroke prevention by aggressive reduction in cholesterol levels (SPARCL) trial. Arch Neurol. 2011;68:1245-51.
64. Ott A, Slooter AJ, Hofman A, van Harskamp F, Witteman JC, Van Broeckhoven C, et al. Smoking and risk of dementia and Alzheimer’s disease in a population-based cohort study: The Rotterdam Study. Lancet. 1998;351:1840-3.
65. Peters R, Poulter R, Warner J, Beckett N, Burch L, Bulpitt C. Smoking, dementia and cognitive decline in the elderly, a systematic review. BMC Geriatr. 2008;8:36.
66. Reitz C, den Heijer T, van Duijn C, Hofman A, Breteler MM. Relation between smoking and risk of dementia and Alzheimer disease: The Rotterdam Study. Neurology. 2007;69:998-1005.
67. Anstey KJ, von Sanden C, Salim A, O’Kearney R. Smoking as a risk factor for dementia and cognitive decline: A meta-analysis of prospective studies. Am J Epidemiol. 2007;166:367-78.
68. Debanne SM, Rowland DY, Riedel TM, Cleves MA. Association of Alzheimer’s disease and smoking: The case for sibling controls. J Am Geriatr Soc. 2000;48:800-6.
69. Doll R, Peto R, Boreham J, Sutherland I. Smoking and dementia in male British doctors: Prospective study. BMJ. 2000;320:1097-102.
70. Hill JW, Futterman R, Duttagupta S, Mastey V, Lloyd JR, Fillit H. Alzheimer’s disease and related dementias increase costs of comorbidities in managed Medicare. Neurology. 2002;58:62-70.