Síndrome de feocromocitoma-paraganglioma de tipo 5 como causa de hipertensión arterial en una paciente colombiana: reporte de caso
Resumen
El feocromocitoma es un tumor derivado de las células de la cresta neural con la capacidad de producir sustancias simpaticomiméticas y, por ende, un cuadro clínico particular. Causa menos del 1 % de los casos de hipertensión arterial sistémica y su incidencia se estima entre 0,4 y 0,6 casos por 100.000 personas cada año, con una supervivencia media de siete años. De todos los tumores sólidos, el feocromocitoma tiene un mayor componente genético, que puede heredarse hasta en el 40 % de los casos. Una vez diagnosticada la enfermedad, se debe definir el tratamiento y el pronóstico, en parte condicionados por las variantes genéticas asociadas, en especial RET, SDHx, VHL y NF1.
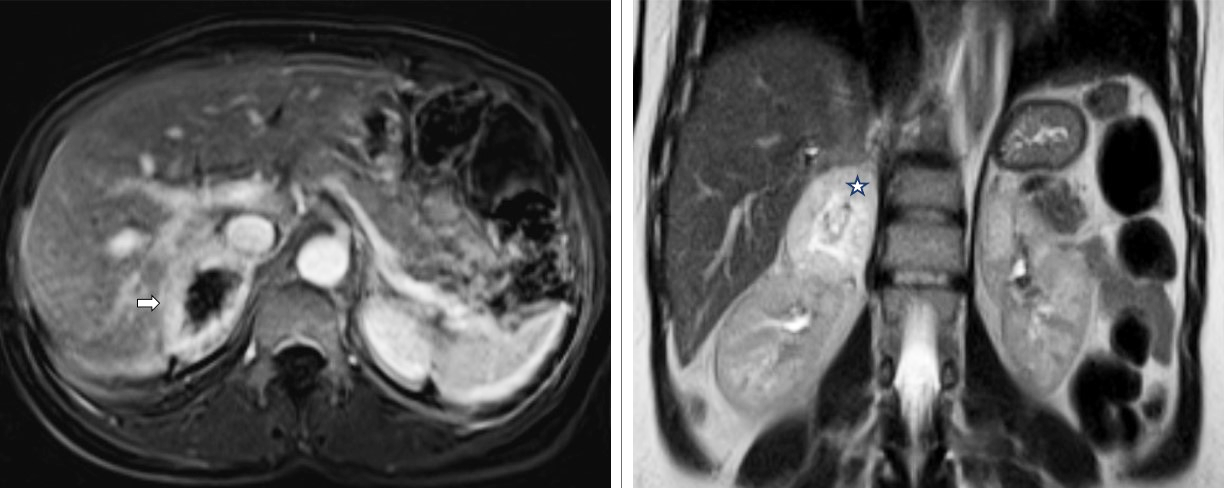
Se presenta el caso de una mujer joven con dolor abdominal e hipertensión arterial sistémica, a quien se le diagnosticó feocromocitoma. Al secuenciar el exoma, se identificó una variante patogénica extremadamente rara y de reciente descubrimiento: SDHA: c.1A>C (p.Met1Leu). La paciente respondió adecuadamente al tratamiento quirúrgico y continuó en seguimiento sin recurrencias.
El abordaje diagnóstico de los pacientes con feocromocitoma comienza con la sospecha clínica, seguida de la medición de determinados metabolitos en sangre y orina, y, finalmente, los estudios de imagenología. Los desarrollos tecnológicos actuales permiten la aplicación de la medicina de precisión en este campo. En este caso de feocromocitoma, se identificó un componente genético importante que no solo afecta al paciente, sino también, a sus familiares. La tamización adecuada del caso índice permite identificar mutaciones y caracterizar mejor la enfermedad.
Descargas
Referencias bibliográficas
Dahia PL. Pheochromocytoma and paraganglioma pathogenesis: Learning from genetic heterogeneity. Nat Rev Cancer. 2014;14:108-19. https://doi.org/10.1038/nrc3648
Berends AM, Buitenwerf E, de Krijger RR, Veeger NJ, van der Horst-Schrivers AN, Links TP, et al. Incidence of pheochromocytoma and sympathetic paraganglioma in the Netherlands: A nationwide study and systematic review. Eur J Intern Med. 2018;51:68-73. https://doi.org/10.1016/j.ejim.2018.01.015
Neumann HP, Young Jr WF, Eng C. Pheochromocytoma and paraganglioma. N Engl J Med. 2019;381:552-65. https://doi.org/10.1056/NEJMra1806651
Navarro EP, Osejo MC, Casas LA, Arango LG, Guzmán G. Experiencia en el manejo de feocromocitoma en los últimos 10 años: serie de casos. Rev Colomb Endocrinol Diabet Metab. 2016;3:33-6. https://doi.org/10.53853/encr.3.3.39
Fränkel F. A case of bilateral, completely latent adrenal tumor and concurrent nephritis with changes in the circulatory system and retinitis. CA Cancer J Clin. 1984;34:93-106. https://doi.org/10.3322/canjclin.34.2.93
Liu Z, Ma J, Jiménez C, Zhang M. Pheochromocytoma: A clinicopathologic and molecular study of 390 cases from a single center. Am J Surg Pathol. 2021;45:1155-65. https://doi.org/10.1097/PAS.0000000000001768
Bausch B, Schiavi F, Ni Y, Welander J, Patocs A, Ngeow J, et al. Clinical characterization of the pheochromocytoma and paraganglioma susceptibility genes SDHA, TMEM127, MAX, and SDHAF2 for gene-informed prevention. JAMA Oncol. 2017;3:1204-12. https://doi.org/10.1001/jamaoncol.2017.0223
Nölting S, Ullrich M, Pietzsch J, Ziegler CG, Eisenhofer G, Grossman A, et al. Current management of pheochromocytoma/paraganglioma: A guide for the practicing clinician in the era of precision medicine. Cancers. 2019;11:1505. https://doi.org/10.3390/cancers11101505
Lenders JW, Duh Q-Y, Eisenhofer G, Gimenez-Roqueplo A-P, Grebe SK, Murad MH, et al. Pheochromocytoma and paraganglioma: An endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014;99:1915-42. https://doi.org/10.1210/jc.2014-1498
Jha A, De Luna K, Balili CA, Millo C, Paraiso CA, Ling A, et al. Clinical, diagnostic, and treatment characteristics of SDHA-related metastatic pheochromocytoma and paraganglioma. Front Oncol. 2019;9:53. https://doi.org/10.3389/fonc.2019.00053
Burnichon N, Brière J-J, Libé R, Vescovo L, Riviere J, Tissier F, et al. SDHA is a tumor suppressor gene causing paraganglioma. Hum Mol Genet. 2010;19:3011-20. https://doi.org/10.1093/hmg/ddq206
Molina L, Salgado J, Amado S. Feocromocitoma y paraganglioma: un reto más allá de la clínica. Rev Colomb Cancerol. 2021;25:3-12. https://doi.org/10.35509/01239015.586
Vasconcelos-Prado OD, López-García AE, García-Montalvo IA. Aspectos genéticos de los feocromocitomas y paragangliomas. J Negat No Posit Results. 2021;6:636-50. https://doi.org/10.19230/jonnpr.4020
Rueda-Galvis MV, Román-Gonzaléz A, Agredo-Delgado V. Síndrome de feocromocitomaparaganglioma tipo 5 asociado a mutación en el complejo de la succinato deshidrogenasa tipo A (SDHA), a propósito de un caso. Iatreia. 2023;36. https://doi.org/10.17533/udea.iatreia.187
Welander J, Garvin S, Bohnmark R, Isaksson L, Wiseman RW, Söderkvist P, et al. Germline SDHA mutation detected by next-generation sequencing in a young index patient with large paraganglioma. J Clin Endocrinol Metab. 2013;98:e1379-80. https://doi.org/10.1210/jc.2013-1963
van Der Tuin K, Mensenkamp AR, Tops CM, Corssmit EP, Dinjens WN, van de Horst-Schrivers AN, et al. Clinical aspects of SDHA-related pheochromocytoma and paraganglioma: A nationwide study. J Clin Endocrinol Metab. 2018;103:438-45. https://doi.org/10.1210/jc.2017-01762
Amar L, Pacak K, Steichen O, Akker SA, Aylwin SJ, Baudin E, et al. International consensus on initial screening and follow-up of asymptomatic SDHx mutation carriers. Nat Rev Endocrinol. 2021;17:435-44. https://doi.org/10.1038/s41574-021-00492-3
Taïeb D, Hicks RJ, Hindié E, Guillet BA, Avram A, Ghedini P, et al. European association of nuclear medicine practice guideline/society of nuclear medicine and molecular imaging procedure standard 2019 for radionuclide imaging of phaeochromocytoma and paraganglioma. Eur J Nucl Med Mol Imaging. 2019;46:2112-37. https://doi.org/10.1007/s00259-019-04398-1
Algunos artículos similares:
- Mario Francisco Guerrero, Elementos para la evaluación eficaz de productos naturales con posibles efectos antihipertensivos , Biomédica: Vol. 29 Núm. 4 (2009)
- Gabriel Bedoya, Jenny García, Patricia Montoya, Winston Rojas, Maria Eugenia Amézquita, Iván Soto, Maria Cecilia López, Jorge Ospina-Duque, Andrés Ruiz-Linares, Análisis de isonimia entre poblaciones del noroeste de Colombia , Biomédica: Vol. 26 Núm. 4 (2006)
- Luis Caraballo, Josefina Zakzuk, Consideraciones sobre la evolución de la respuesta inmunitaria Th2 y sus posibles relaciones con parasitosis y alergia , Biomédica: Vol. 32 Núm. 1 (2012)
- Jefferson Antonio Buendía, Actitudes, conocimientos y creencias del paciente hipertenso sobre la medicación antihipertensiva , Biomédica: Vol. 32 Núm. 4 (2012)
- Lisseth Cabarcas, Eugenia Espinosa, Harvy Velasco, Etiología del retardo mental en la infancia: experiencia en dos centros de tercer nivel , Biomédica: Vol. 33 Núm. 3 (2013)
- Juan J. Yunis, Luis E. Acevedo, David S. Campo, Emilio J. Yunis, Origen geno-geográfico de haplotipos STR del cromosoma Y en una muestra caucásico-mestiza y afrodescendiente de Colombia , Biomédica: Vol. 33 Núm. 3 (2013)
- Carlos A. Isaza, Franciso J. Osorio, Giovanny Mesa, Juan C. Moncada, Patrones de uso de antihipertensivos en 11.947 pacientes colombianos. , Biomédica: Vol. 22 Núm. 4 (2002)
- Carlos Alberto Palacio, Jenny García, María Patricia Arbeláez, Ricardo Sánchez, Beatriz Aguirre, Isabel Cristina Garcés, Gabriel Jaime Montoya, Juliana Gómez, Angela Agudelo, Carlos Alberto López, Jorge Julián Calle, Carlos Alberto Cardeño, Juan Fernando Cano, María Cecilia López, Patricia Montoya, Claudia Patricia Herrera, Natalia González, Alejandro González, Gabriel Bedoya, Andrés Ruiz, Jorge Ospina, Validación de la entrevista diagnóstica para estudios genéticos (DIGS) en Colombia. , Biomédica: Vol. 24 Núm. 1 (2004)
- Nora Alejandra Zuluaga, Jorge Mauricio Cuartas, Juan Guillermo Londoño, Genética de la preeclampsia: una aproximación a los estudios de ligamiento genético. , Biomédica: Vol. 24 Núm. 2 (2004)
- Diana María Valencia, Carlos Andrés Naranjo, María Victoria Parra, María Antonieta Caro, Ana Victoria Valencia, Carlos José Jaramillo, Gabriel Bedoya, Asociación y efectos de interacción en los genes AGT, AGTR1, ACE, ADRB2, DRD1, ADD1, ADD2, ATP2B1, TBXA2R y PTGS2 sobre la hipertensión en la población antioqueña , Biomédica: Vol. 33 Núm. 4 (2013)
Derechos de autor 2024 Biomédica

Esta obra está bajo una licencia internacional Creative Commons Atribución 4.0.
| Estadísticas de artículo | |
|---|---|
| Vistas de resúmenes | |
| Vistas de PDF | |
| Descargas de PDF | |
| Vistas de HTML | |
| Otras vistas | |